您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-09-28 13:30
前言
随着液相的普及,紫外可见分光光度计(UV)的使用频率在逐渐下降。十年前,UV大量用于溶出度和含量的测定。尤其是溶出度的测定,药典中收载了大量用UV测定溶出量的品种。目前,UV大都仅用作定性鉴别了,但其在某些特殊情况下仍有着不可替代的作用。
UV测定的优缺点:
如有辅料或是其他组分的存在,UV在某个波长下测定的是组分的叠加信号,不能区分开,而HPLC却能很好地将辅料或其他干扰组分在色谱柱上分离开,这种定量测定专属性差的缺点制约了UV的发展。但是正是这个缺点有时可能成为优点,例如利用这个“优点”可测定易降解化合物的叠加响应来计算药物的溶出度。
此外,一些溶出仪与UV联用在线测定溶出量,甚至诞生了光纤溶出仪;UV测定的成本相对HPLC也低很多。因此,UV还是有它用武之地的。在利用UV进行固体制剂溶出度和含量的定量测定时,辅料的干扰成了难以绕开的障碍。本文就笔者的相关经历并结合理论和文献,谈一谈UV测定时消除辅料干扰的一些技巧。
1绕开辅料吸收较强的波段
这是UV测定时消除辅料干扰的最简易方法,如能用此法解决干扰问题,那最省事了。具体步骤如下:
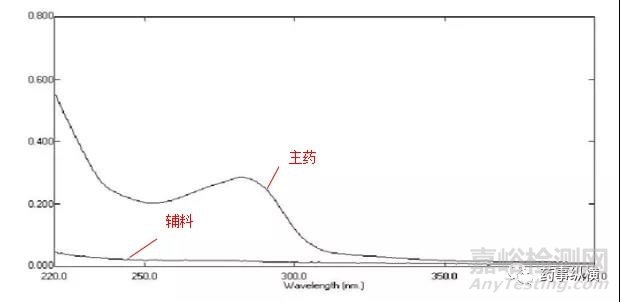
分别将制剂中处方比例的主药和空白辅料配制成合适浓度的溶液,在一定波长范围内(如200nm~400nm)扫描得光谱图。
查看辅料和主药的光谱图,一般辅料在低波段的吸收均较大(250nm之前),从此波长往后找,看能否找到辅料的吸收几乎没有(贴近零线)而主药的吸收仍然较大的波长。如果此波长是主药的吸收峰,那更好了。如果不是吸收峰,较平坦处也行。前两者都不是,在陡坡处的波长也能勉强测定,现在UV仪的波长精度较好,误差不会太大。
一般溶出度测定时的辅料干扰控制在2%以内即可,当然越逼近零越好。
在此,顺便说一下,为何定量测定时要选择最大吸收波长处,HPLC也是如此,因为在最大吸收波长处,吸收最大且光谱较光滑平坦,斜率几乎为零,若因波长的精度问题轻微波动,不会引起吸收度的较大误差。而在陡坡处,斜率较大,波长的轻微误差就会引起吸收度的较大变化。不过现在的UV仪器,波长准确度能控制在0.2nm左右,这种波长误差引起的吸收度误差大可不必担心。
2双波长等吸收差值法
此法需要满足一定的条件才适用,具体条件和步骤如下:
分别将制剂中处方比例的主药和空白辅料配制成合适浓度的溶液,在一定波长范围内(如200nm~400nm)扫描得光谱图。
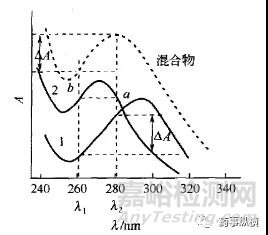
查看主药和空白辅料的光谱图。如主药在λ2有较大吸收,吸收度为A1(λ2),在此波长处辅料也有吸收,为A2(λ2)。
查看辅料的光谱图。辅料光谱图上有无与A2(λ2)等吸收的波长,如有,假设为A2(λ1),而且在此波长处主药几乎没有吸收。
测定时,采用双波长测定,λ1和λ2,计算两个波长处的差值。假设在λ2处主药和辅料的总吸收度为A(λ2)。
A(λ2)= A1(λ2)+ A2(λ2)
其中A2(λ2)= A2(λ1),代入上式
A(λ2)= A1(λ2)+ A2(λ1)
A(λ2)- A2(λ1)=A1(λ2)
如此,求得的差值A(λ2)- A2(λ1)就是主药在λ2处的吸收度A1(λ2)。此法测定必须符合两个基本条件:1)干扰组分辅料在这两个波长应具有相同的吸收度;2)待测组分在这两个波长处的吸收度差值应足够大,即主药在λ2处的吸收度较大,而在λ1处几乎没有吸收。大部分品牌的UV工作站都能进行双波长测定,如岛津的UVprobe和安捷伦的21CFR紫外工作站,有的还能直接读出双波长间的差值。所以如能满足上述条件,测定过程也相当简便,主要是能很好地消除辅料的干扰。下图虽不是实际案例,但通过此示意图能方便理解。
3导数光谱法
此法听起来似乎很玄乎,其实理解之后,也并不复杂,笔者曾经接触过这样的案例,如某片进口注册标准中的溶出度测定就是用的导数光谱法,其目的是利用导数光谱法消除片中的辅料干扰。导数光谱的定量原理:

从上式一阶导函数中,可以看出一阶导数与样品浓度仍然成线性关系。同理,二阶导数和三阶导数等高价导数也与样品浓度成线性关系。注意,这里是对光谱函数的导数,得到导数函数,而不是吸收度的导数,吸收度值是一常数,求导后为零。
利用导数光谱法消除辅料干扰的要求和步骤如下:
分别将制剂中处方比例的主药和空白辅料配制成合适浓度的溶液,在一定波长范围内(如200nm~400nm)扫描得光谱图。
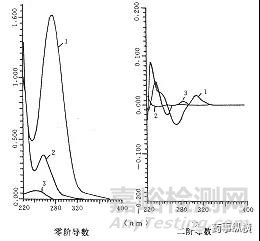
查看主药和空白辅料的光谱图。将这两个光谱图分别转换成导数光谱图,岛津的UVprobe和安捷伦的21CFR紫外工作站都有此功能。在转换导数光谱时需要设置Δλ值,该值可选择默认值也可根据需要选择合适值。
查看主药和辅料光谱图经转换后的导数光谱图。对应于主药导数光谱图中的波峰或波谷处,辅料导数光谱图是否几乎没有吸收(越贴近零线越好),一般情况下,辅料光谱图经一阶导数转换后,原来轻微的干扰会消失殆尽。如一阶导数后,辅料还是有较大干扰,可再经二阶导数或三阶导数转换。
定量测定时,采用导数光谱上适宜的振幅作为定量参数,而不再称作吸收度了。常用的方法有峰谷法和峰零法。峰谷法是测量两相邻峰谷之间的垂直距离,峰零法是峰或者谷到零线之间的垂直距离。
方法确定后,在正式测定进行光谱扫描时,扫描的波长范围可适当缩小,如250nm~300nm,这样可节约扫描时间。
结语
上述提及的几种方式,可根据实际需要选择,能简则简,解决干扰问题就行。其实还有其他更为复杂的方式,没有必要进一步折腾。UV法如用得好用得熟练,能大大节约成本,有时还能弥补HPLC的不足。当然,随着HPLC和UPLC的快速发展,UV法只能作为配角了,主角和配角都使用恰当,事情会做得更好。本文仅代表个人观点,如有不当,还请各位同仁多多包涵。
参考文献
[1] 沈春鸣. 双波长分光光度法测定维C银翘片溶出速率[J]. 中国药业,2003,12(9):35-36.
[2] 朱亚尔,王华,景文涛,等. 复方磺胺甲噁唑分散片溶出度方法学研究[J]. 分析试验室,2003,22(11):264-266.
[3] http://www.gbw114.com/news/n35693.html
[4] 冀宛丽,赵家太. 一阶导数光谱法测定氯霉素滴眼液中氯霉素的含量[J]. 中国现代应用药学杂志,2008,25(8):723-724.
[5] 臧志和,陈代勇,辛志伟,等. 一阶导数光谱法测定氧氟沙星片的含量[J]. 中国抗生素杂志,2001,26(3):230-231.
[6] 夏曙辉,陆崟,曹凌燕,等. 二阶导数光谱法测定盐酸利多卡因胶浆中盐酸利多卡因的含量[J]. 中国医院药学杂志,2007,27(4):564-565.
[7] 全山丛,石力夫,张德莉,等. 二阶导数光谱法测定地塞米松霜中氯霉素的含量[J]. 中国医院药学杂志,1998,18(1).
来源:Internet


