您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-01-09 18:10
日常药物测定的准确性通常是通过控制分析误差来实现,其所采用的测定方法是否准确必须进行分析方法验证来判断,因此分析方法验证在现代质量控制中意义极其重大,避免了因方法问题导致不合格药品流入市场,也是保证药品质量可控制性的重要一环,分析方法验证体现企业技术水平,同时验证的结果对以后日常测定出现问题的解决有很大帮助,此外,方法验证对于企业的新药报批、国外的注册和GMP检查均非常关键。
中国药典2020版征求意见稿[1]、美国药典[2]及ICH[3]分析方法验证都制定有相应的指导原则,但均很宽泛,缺少具体操作的描述,也没有说明规定的理由,日常验证中弄不清技术要求和法规要求的区别,或过于严格,或为了应付检查而做的过于简单,或按照自己浅显的理解做错,如仅仅通过信噪比即确定为方法的定量限,把线性的范围等同于定量的范围,系统适用性参数的制定不是根据验证结果来制定,有关物质的精密度测定的样品使用不含有待测有关物质等问题。
分析方法的好坏是开发出来的,而不是验证出来的,方法的好坏决定验证工作的顺利与否,所以方法开发非常重要,开发时应充分考虑到分析方法验证的风险点和方法的经济性,有人包括分析方法的耐用性需要考虑到以后日常测定批次多,次数多的异常发生率的问题,特别样品的预处理过程是否科学?往往通过后来的验证无法鉴别,需要在开发过程中加以研究如衍生化条件、样品溶解处理的方式和条件等,一旦验证没有识别出方法本身的问题,在以后日常测定出现异常或OOS,给质量管理带来非常大的麻烦。
1、关于分析方法生命周期问题:
近来,药监部门加强了对分析方法的生命周期 [4]管理意识,ICH和美国药典论坛纳入了包括分析方法生命周期管理的新的理念,特别美国药典拟将一个新的通用章节(1220)“分析方法的生命周期”合并于美国药典中。
分析方法的生命周期分三个阶段,即设计和开发阶段、验证阶段和和持续监测阶段,分析方法的好坏是开发出来,而不是验证出来的,所以,分析方法的验证不是孤立的一项工作,和分析方法的开发甚至和分析方法的设计分不开的,分析方法开发和验证是一个有机整体,即使分析方法验证完成后,需要在其生命周期中用于常规的放行或稳定性测试中,持续监测方法的性能,监测该方法的测定结果是否满足质量控制要求,这是分析方法验证的最后一阶段,直至分析方法的终止,这和产品持续稳定性考察相同原理,所以企业年度报告中建议增加一项分析方法的性能总结等内容,如分析一年共有多少OOS(超标)和OOT(超趋势)是由于分析方法本身的缺陷引起,并制订纠正预防措施对所采用的分析方法进行持续改进,目前国内几乎没有企业进行这项工作,法规检查也忽略了这一重要内容。
2、关于分析方法的专属性
2.1 峰纯度问题:专属性不好往往会导致测定的系统误差,而且在日常测定很难发现,特别多个组分出峰完全重叠的情形,这些只有通过验证专属性的峰纯度加以考察,对于老产品的专属性的验证使用过期样品进行峰纯度测定可能比强制降解样品更为科学,此外需要注意实际验证中经常出现峰纯度“假合格”和“假不合格”现象,“假合格”主要由于检测技术的限制,但“假不合格”情况更多,主要是样品响应过高或过低,波长选择涵盖或接近流动相有机相的截止波长,缓冲盐的浓度过高或流动相附加剂的纯度影响等原因,所以杂质很难准确也没有必要进行峰纯度的测定。
2.2 物料平衡问题:强制降解的物料平衡是广大分析工作者极为头痛的问题之一,由于物料平衡主要是考察分析方法能否将所有杂质有效检出,所以含量测定没有必要进行物料平衡的考察,物料平衡的接受范围问题,至今没有统一的规定,其实也很难统一,需要具体情况具体分析,根据样品中各组份性质、降解率大小、强制降解杂质的结构、质量标准中杂质限度要求等因素综合考虑,建议为95.0~105.0%较为合适,有些企业制定为98.0%~102.0%,只考虑HPLC方法本身误差,没有考虑未知杂质的结构、介质pH等因素对测定的影响,接受标准过于严格,有些企业对于制剂考虑到辅料影响,制定为90.0%~110.0%,又过于宽松,失去物料平衡考察的意义。
2.3 分离度的计算问题:分离度是HPLC测定中重要的系统适用性参数,也是专属性重要的考察参数之一,对于拖尾峰和峰形不好的峰,现代的色谱积分技术基本均能够很好解决,对于分离不好,尤其是色谱峰峰响应差别大的两组份不完全分离的情形,现代积分技术很难科学解决,改善分离成为唯一手段。中国药典2020版征求意见稿[5]和美国药典[6]均有二种方法即峰底宽法和半峰宽法,其实二者本质均一样,欧洲药典[7]有两种计算方法,即半峰宽法和峰谷比法(p/v法)。但对于二个组分响应相差很大或一个组分由于过载产生严重变形时,这些传统的分离度公式均不适用,会导致结果偏低或偏高,不能有效的反应真正的分离情况,对于这个情况如采用峰谷比法[7],或使用柱效、选择性和容量因子的公式进行计算[8]更为科学,可惜各国药典均没有收载这一科学的分离度计算方式。
2.4 空白辅料峰的问题:
空白辅料虽通过过滤使溶液澄清,但其在测定溶液中浓度往往较大,通常是主成分浓度的几倍甚至几百倍以上,在进样后使流动相体系产生瞬间的波动,导致在死时间附近出现辅料峰,或辅料的折、散射而产生峰,但这并不是辅料吸收峰,所以其响应并不遵循朗伯比尔定律,有时同样配方量的空白辅料和空白片产生的峰面积相差很大,不同品牌的辅料甚至同一品牌不同贮存条件和时间产生的峰也不一致,鉴于辅料峰的复杂性以及工艺的保密性,实际工作中如出现辅料产生的峰,一定要进行详细的调查,弄清是什么辅料什么原因产生的,并根据实际在方法中确定处理方式,而不能简单的在检验规程中要求进样空白辅料溶液,这样既不科学也不现实。
3、关于分析方法验证的精密度
实际测定时虽然精密度好,但由于方法或测定仪器等系统误差因素,往往可能准确度未必好,但反之,精密度不好准确度很难得到保证,因为测定的准确性是建立在良好的精密度基础上,所以日常测定是通过测定平行样品,考察测定误差来达到控制准确度的目的,验证精密度过程中常见以下一些问题,如:
3.1 在有关物质和残留溶剂的精密度测定时,当使用样品测定结果为未检出,由于分析方法是适用于符合质量标准或不符合质量标准的所有样品,所以应该在样品中加入适量的(如限度的50%~100%)待测杂质对照品,测定其含量的RSD%考察精密度是否符合测定要求,如不是同一贮备液进行稀释,由于贮备液取样量不同,需要通过计算回收率的RSD%来考察精密度是否符合测定要求。
此外,虽然质量标准中没有对杂质个数进行限定,但在有关物质的测定方法精密度验证时除考察其含量的精密度外,还要考察忽略限以上杂质个数是否一致,这往往为大家所忽视,特别质量标准中如规定最大未知单一杂质时,需要标明最大单一杂质的保留时间或相对保留时间,防止不同样品测定时最大单一杂质的峰发生改变而失去统计的意义。
3.2 对于使用含量均匀度测定结果作为含量放行数据的分析方法,精密度的验证相对比较复杂,由于含量均匀度一般取10片测定结果的平均值作为含量放行数据,同时计算A+2.2S值作为均匀度考察的指标,在该情况下,含量的精密度验证应该按照含量均匀度的10份而不是6份测定结果进行精密度考察,即每人测定10份,二人共20份,计算A+2.2S值作为考察指标,并和同批通过研磨等手段取得均匀样本测定的含量进行比较,以考察样品处理方式不同对结果的影响。
3.3 精密度的接受标准问题:中国药典2020版征求意见稿[1]有详细规定,但这些指标值只考虑浓度对误差的影响,没有考虑方法操作(如预处理过程)、仪器本身等影响因素,但作为药典有标准总比没有标准好,药典也规定了适当放宽情况,所以可以根据实际情况制定接受标准,如对于含量RSD%接受标准一般为≤2.0%,但这也不是绝对的,对于原料药或范围要求很窄的制剂(制剂含量范围主要是考虑生产和工艺的波动范围,适当考虑分析测定的误差范围),应该制定更小的RSD%接受标准如≤1.0%,以保证方法能够满足质量控制的要求,对于杂质测定方法验证的精密度接受标准列下表1供验证时参照使用,此表也适用于残留溶剂等微量成分的定量测定。
表1:杂质测定方法精密度接受标准推荐表
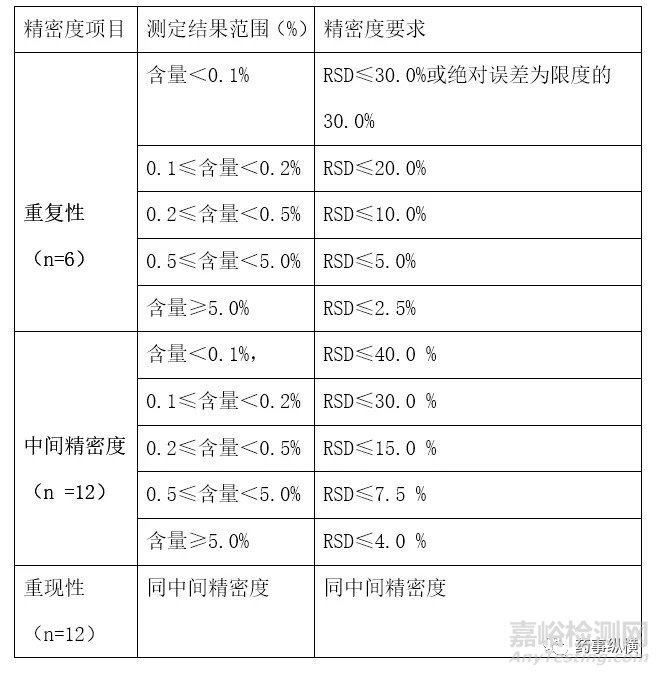
4、关于分析方法验证的检测限和定量限
任何测定方法包括测定仪器都有灵敏度的限制,对于微量组分的测定,考察检测限和定量限非常必要,对于低于检测限的测定结果应为未检出(Not detected或ND表示),对于大于检测限但低于定量限的测定结果,建议结果表达为低于定量限(Below Quantitation Limit或BQL),只有高于定量限才可能也必须报告具体检测数据,对于小于忽略限的测定结果通常使用低于忽略限(Below Disregard Limit或BDL),但需要注明忽略限的具体规定如0.03%,此外忽略限不能小于定量限,关于检测限和定量限需要注意以下几个问题:4 关于分析方法验证的检测限和定量限
4.1 信噪比法的缺陷:ICH[3]信噪比法测定检测限和定量限,是将仪器的灵敏度测定方法用于分析方法,不太科学,因为待测组分和噪音的响应受仪器、柱效、试剂等影响,信噪比法测定的检测限和定量限不具有普遍意义上的代表性,只是一个偶然值,根据定量限原理,信噪比不小于10,必须所有测定结果(如通常6次)均不小于10,不能取平均值不小于10,也没有必要制订一个范围如8~12,此外对所有定量限下测定峰面积的偏差需要制定一个接受标准如RSD%≤10%以弥补精密度测定没有考察定量限浓度下的精密度,同时定量限浓度需要符合线性和准确度要求,只有这几个条件全部具备才能确定为定量限,为此,通常先根据质量控制要求确定一个忽略限,然后直接进行信噪比测定,关注信噪比是否不小于10而不必考虑信噪比的具体数值是多少。
由于信噪比法测定定量限值影响因素多,波动大,所以定量限除考察信噪比外,还应制订定量限不大于杂质限度的50%的要求以弥补这一不足,也是准确度测定浓度为限度50%、100%和150%的考虑,如果定量限接近限度值,建议使用限度法进行杂质控制而不能使用定量法进行杂质控制。
基于响应值标准偏差和标准曲线斜率法虽然和信噪比法原理相同,但比信噪比法结果重现性更好些,现在有代替信噪比法的趋势。
4.2 关于噪音的测定:
噪音的测定非常讲究,除应该选择平滑的地方外,建议应该在离目标峰附近前后两段进行,取其最高者,因为只有目标峰附近才影响目标物的测定,远离目标峰噪音对待测组分测定几乎没有影响。测定噪音的时间30秒,如果成份多,梯度洗脱的方法,15秒即可,因为15秒两组分已能得到很好分离,目前所有组分用某一段相同的基线测噪音并不太科学(除非基线很一致),收集60秒以上更没必要,是自找麻烦的做法。
4.3 忽略限制定问题
忽略限和报告限数值相同但含义不同,制定忽略限需要根据方法和测定质量控制的要求以及法规要求等因素综合考虑,由于仪器的噪音等因素,使用HPLC测定杂质时,虽然中国药典没有提及忽略限,但实际工作中制定忽略限,将小于忽略限的峰不予计算,非常科学, ICH对报告限有具体规定,但只是基本要求,建议日常工作中制定更为严格容易获得审评专家的认可,通常忽略限一般规定为0.03%,但和其他技术指标一样不能一概而论,具体制定策略需要根据方法本身的噪音大小,杂质的限度大小等因素来制订,原则上忽略限为杂质限度的1/5以下,如1/5计算值大于0.03%,取0.03%,如小于0.03%取1/5计算值,而且忽略限必须不能小于定量限,对于稳定性考察的测定由于需要进行趋势分析,建议直接将定量限作为报告限更有适用性。
4.4 关于检测限的问题:法规上对于杂质含量的检测限是否进行要求根据具体情况来定,这个需要和以后的放行测定的样品结果来分析这一问题,如果该杂质在样品中未检出,需要进行检测限的测定,否则无法知悉未检出的判定,如果产品的杂质不可能出现未检出的情形,可以不做检测限,对将来样品质量情况的预测是很难的,所以进行检测限的测定是很明智的选择。
5、关于分析方法验证线性和范围
线性关系考察是定量分析的基础和必要条件,定量是基于呈一元一次方程的线性关系进行的,对于不呈线性的方法,可以通过数学转换如HPLC-ELSD检测或使用二次拟合曲线法如HPLC-CAD检测,但线性不是定量的充分条件,呈线性未必能够准确定量,特别低浓度由于辅料干扰(线性一般只使用对照品进行)以及背景吸收和扭曲效应等因素的影响,所以需要考察其准确度和精密度,确定测定范围,此外除了考察相关系数以确定是否成线性外,还必须考察回归方程的截距问题,通常使用两种方法,一种为Y轴截距绝对值与标准(100%)之比法,另一种为剩余标准差标准(100%)之比法,二种方法均能反应截距的大小,对于含量应该不大于2%,对于有关物质和残留溶剂应该不大于25%(建议不大于15%更科学),如果截距不符合要求,不说明方法不能定量,只是不能采用常用的一个对照浓度进行测定,需要采用多个浓度的标准曲线法进行。
中国药典2020版征求意见稿[1]参照ICH要求报告残差平方和,这一指标是考察线性的各点偏离程度,是综合性考察线性斜率、截距、线性的指标,无法制定统一可接受标准,法规上通常是要求报告残差平方和数值用于评定方法的优劣。
6、关于分析方法验证的准确度
虽然检测永远不能测定到待测成分的真实含量,但测定结果尽量接近真值是分析工作者和分析方法的最高目标,虽然加样回收率对于某些样品由于崩解溶出等因素的影响不能完全反映准确度情况,但仍然不失为验证方法准确度最好的方法,了解以下几个问题对于准确度的测定会有大有好处。
6.1 中国药典2020版征求意见稿[1]关于回收率的接受标准范围是非对称范围,如98~101%等,因为不可能有化合物含量超过100%,超过100%部分完全是分析误差,所以原料药的标准范围呈非对称如98.5%~101.0%,而加样回收率则不同,真实含量为100%的上下波动呈对称的正态误差分布,这一规定容易产生误导,即分析方法允许存在系统误差,即高于100%的误差小于低于100%的误差,而且回收率范围小数点位数不科学,98~101%应为98.0~101.0%,此外还有存在其他诸多问题,建议实际工作不予采纳,对于杂质测定方法验证的回收率范围及误差接受标准列下表2供验证时参照使用,此表也适用于残留溶剂等微量成分的定量测定。
表2杂质回收率范围及误差表
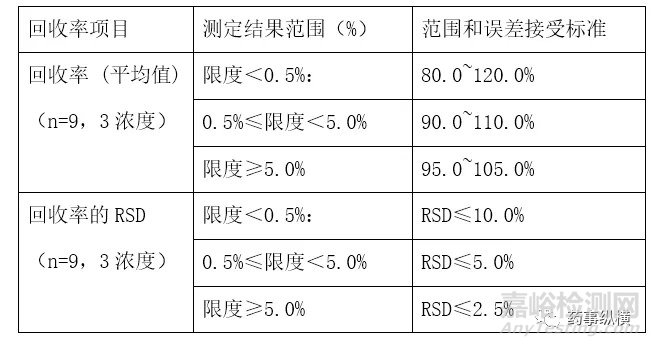
6.2 对于有关物质(包括溶剂残留)的定量测定,由于质量标准规定不是范围,仅是限度,所以应该分二部分进行,即LOQ的浓度下进行和限度附近(通常为限度浓度的50%、100%和150%),其中限度浓度是决定产品合格不合格的关键浓度,是风险最大的区域,所以需要在该浓度的附近进行准确度的测定,因此有些企业所有情况均不进行50%的限度浓度是不科学做法,回收率范围一般规定为80.0%~120.0%,九次测定结果的RSD应≤10.0%,如果限度浓度高可以严格到90.0%~110.0%,甚至95.0%~105.0%,而LOQ浓度比较低加上往往远离限度值,风险较小,回收率范围可以适当放大到70%-130%,此外,根据验证回收率来制订内控质量标准不失为一种科学的方法,如回收率为85.0%,考虑方法的准确度,限度可以考虑从0.2%修订为0.17%,弥补测定的误差。
6.3 对于有关物质测定,由于杂质对照品较难获得,常常使用与主成分相比进行测定,由于杂质和主成分的结构不同,响应可能不同,需要进行校正才能测定真实含量,否则为表观含量,中国药典2020版征求意见稿[5]和欧洲药典[7]使用的为校正因子,而美国药典[6]使用的是响应因子,二者互为倒数关系,对于质量型检测器由于响应与待测组份的质量关系,所以通常不需要测定校正因子。6.2对于有关物质(包括溶剂残留)的定量测定,由于质量标准规定不是范围,仅是限度,所以应该分二部分进行,即LOQ的浓度下进行和限度附近(通常为限度浓度的50%、100%和150%),其中限度浓度是决定产品合格不合格的关键浓度,是风险最大的区域,所以需要在该浓度的附近进行准确度的测定,因此有些企业所有情况均不进行50%的限度浓度是不科学做法,回收率范围一般规定为80.0%~120.0%,九次测定结果的RSD应≤10.0%,如果限度浓度高可以严格到90.0%~110.0%,甚至95.0%~105.0%,而LOQ浓度比较低加上往往远离限度值,风险较小,回收率范围可以适当放大到70%-130%,此外,根据验证回收率来制订内控质量标准不失为一种科学的方法,如回收率为85.0%,考虑方法的准确度,限度可以考虑从0.2%修订为0.17%,弥补测定的误差。
校正因子最好为不同仪器和人员至少测定两次,允许误差一般为10%,这是基于杂质回收率要求在90.0%~110.0%,其依据为国家食品药品监督管理局药品审评中心[9]规定0.9~1.1可不需要校正的规定(英国药典要求将0.8~1.2可不进行校正的规定范围过于宽松,建议不予采用),此外,如药典方法进行验证或确认时,和药典规定值比较误差也应在10%内,由于影响校正因子准确的因素很多如对照品含量误差(如吸水)、保留时间差别大、峰型差别大(如拖尾严重)、DAD和紫外检测器的差别、盐类药物对照品没有折算成活性部分等原因,鉴于药典校正因子的测定次数多,日常测定尽量使用药典的校正因子进行。
即使校正因子在0.9~1.0之间,建议按照实际测定校正因子的数值进行,可避免多次测定结果值在边缘上下的时候处理数据的麻烦,如校正因子发生改变,必须对以前测定的数据进行评估,特别是校正因子变大的情形。鉴于校正因子重要性,不管结果是否小于1.0,其结果建议均应保留小数点二位。
杂质的校正因子的测定有多种方法,但使用杂质线性的斜率和主成分的线性斜率比最为常见,杂质的线性最大浓度为限度的150%(或120%浓度),最低浓度为定量限(或忽略限浓度),但主成分的线性浓度要考虑不同限度要求的杂质以及低浓度的因素,最大浓度的确定比较复杂,建议使用最大的限度值的杂质的150%或测定的对照低浓度的150%(二者取最大者)进行,以满足所有杂质测定要求。
校正因子如果不在0.2~5.0之间,会带来分析误差的放大或减少效应,测定数据不可靠,建议修改检测器或检测波长,如有难度,改为外标法。
6.4 虽然本文主要针对HPLC的测定方法,但顶空气相色谱准确度的验证值得关注,由于其测定原理是气液平衡,测定的是相对回收率,所以影响组份气液平衡的因素在方法开发和方法验证中均应考虑,特别是对照溶液和样品溶液的溶质差别,样品溶液中含有高浓度的样品,存在样品的基质效应,该效应有时是正效应,有时是负效应,正效应导致回收率偏高,负效应导致回收率偏低,具体与待测组份的结构、沸点、性质、稀释剂的种类有关,基质效应是顶空最大难题,在满足测定要求情况下减少样品量可以减少基质效应,但不能彻底消除,彻底消除基质效应的最好方法是采用标准加入法进行测定。
6.5 制剂和原料药的主成分含量的准确度测定的浓度选择范围均在80%、100%、120%三个浓度下进行,但其目的并不相同,制剂的标示量通常在90.0%~110.0%的范围,考虑到风险问题以及方法适用性更广,所以进行80.0%-120.0%浓度范围进行,而原料药是基于药典精密称量约的含义为±10%,即供试品浓度的90.0%~110.0%,同样考虑到风险问题以及方法适用性更广的原因,适当放宽范围为80.0%~120.0%浓度范围,对于中药,由于中药的主成分范围比较宽,通常放宽到50%~150%的浓度范围。
中国药典2020版征求意见稿[1]规定如不能得到辅料全部成分,使用样品加入待测对照成分进行回收率考察,这样操作会导致实际待测成分均在测定浓度之上,没有包含低于测定浓度的准确度情况,低于标准规定附近的边缘数据没有进行准确度验证,存在极大质量控制风险,所以对于企业来说,知晓处方和工艺情况下,最好模拟处方进行回收率试验。
7、关于分析方法验证的耐用性
分析用仪器虽然经过校正,但允许存在一定的误差,如紫外检测器波长允许±1nm,柱温允许±1~2℃等,分析方法应该适用于所有经过校正合格的仪器上都能准确测定。此外,进行耐用新测定另外一个目的是通过耐用性测定后,在耐用性变动参数范围内改变均不算改变方法,比如流速,虽然仪器流速误差小,但仍然进行有±10~20%的变动耐用性考察,主要是实际测定时为了保证符合系统适用性要求对某些参数进行调节,此外,耐用性还能发现方法的风险点,为以后日常测定出现异常提供一些参考的信息。
7.1 耐用性除测定系统适用性参数外,还需要测定目标物的含量并和正常测定条件进行比较,在规定的范围内,对于杂质测定方法还需要统计杂质个数和最大未知杂质的保留时间或相对保留时间以考察不同条件下的方法的检测能力是否耐用。
7.2 关于耐用性波长的考察,中国药典2020版征求意见稿[1]没有提及,主要是否进行波长微小变化的考察取决于测定方法,如HPLC法采用紫外检测器采用外标法测定时,由于对照品和待测定成分结构相同,其紫外吸收相同,所以耐用性没有必要进行不同波长下的耐用性考察,但如杂质为与主成分比较如自身对照法测定杂质,由于杂质和主成分结构不同,不同波长的校正因子可能不同,而不同仪器波长存在一定的偏差如±1nm,所以即使中国药典2020版征求意见稿[5]规定分析方法的波长不能改变,但耐用性验证需要考察不同波长情况下的测定结果以保证分析方法适用于所有校验合格的仪器。
7.3 耐用性参数的变化在考虑仪器和计量误差范围外适当放宽即可,如果变化值放得过宽,会引起耐用性失败,如柱温,通常柱温箱的温度精度为±2℃,耐用性只需做到±3℃即可,同样波长变化范围±2nm(或±3nm)。耐用性结果如发现分析条件对测定结果影响很大,必须在方法中予以说明,如缓冲液pH的变化范围±0.2,出现耐用性不好,可以严格到±0.1,但必须在分析方法中注明(不注明可视为±0.2)。
7.4 对照品溶液和样品溶液稳定性考察可以在耐用性中进行,因为对照品溶液需要作为控制对照在检测最后测定,以考察系统是否漂移。而样品溶液需要在稳定性情况下才具有代表性,测定时仪器不关闭才有验证意义,这和贮备液的稳定性考察不同,后者不是方法本身,是为了节省对照品的用量而采取的措施,所以不需要在方法验证中同时进行,作为企业的质量管理工作,单独进行,切不可混淆。
8 、关于分析方法验证的系统适用性
系统适用性是保证测定结果准确的前提,分析方法验证不管进行什么项目的验证,均应进行系统适用性的测定以考察测定是否有效?如果测定方法中对照溶液参与测定计算还应进行检查对照的测定以考察工作对照的容量操作是否有问题?如测定时间较长,还应该进行控制对照的测定,以考察整个测定过程中系统是否漂移?如果方法比较稳定,不必在测定过程中定期测定控制对照,此外,在验证总结中需将所有的系统适用性的数据均予以列出,以有利于评审专家的审评。
9、方法确认和方法转移
分析方法确认和分析方法转移是一个比较经济的验证分析方法准确性的方式,中国药典2020版征求意见稿[10-11]专门增加了分析方法确认和分析方法转移两个指导原则,从法规上给予分析方法确认和分析方法转移合法地位,是新版药典的亮点之一,虽然指导原则比较原则,但其指导意义很大,如制剂企业购买的原料药辅料,其验证工作量巨大,如采用分析方法转移的方式可大大减轻企业负担,方法转移可以同一公司不同部门,也可以不同公司之间,但产品的生产工艺或处方必须完全一致,否则只能验证或确认,转移方式有四种[11],即比对试验、共同验证、再验证、转移豁免四种方法,其中比较性测试最为常用。
方法确认药典[10]指导原则没有作出具体规定,需要根据具体情况经过科学评估进行选择,由于原料药没有辅料的干扰,也不存在崩解等复杂因素,对于药典方法原料药采取确认是一个既经济又科学的方式,确认一般进行专属性、精密度和线性范围三个项目,分别考察干扰情况、测定误差和定量基础是否影响测定方法的准确度,如系杂质等微量组分的测定方法,还需要进行定量限和检测限的测定以考察方法灵敏度,对于制剂测定方法,由于辅料及工艺对测定方法准确度影响较为复杂,建议进行验证,即使确认,项目也要经过评估,通常除了耐用性的其他所有验证项目。
10、其他
10.1 对照品标化所采用分析方法应该经过验证,但验证需要标化后的对照品才能进行,这就存在先有鸡还是先有蛋的矛盾,如何解决这一问题,建议将对照品的标化和分析方法验证一起进行,二者同时进行批准以解决这一矛盾。
10.2 检测限和定量限、线性和范围、定量限浓度下的准确度三个项目一般直接使用对照品进行,也不需要加入样品,而其他项目则需要具体项目具体分析,如原料药不含辅料,准确度的测定时使用50%浓度的样品溶液,分别加入30%、50%、70%的测定浓度的对照品来测定回收率。
10.3 通常分析方法和工艺及设备验证不同,不需要进行周期性再验
证,即使仪器搬迁也不需要进行方法本身的再验证(仪器再确认是必须的),这是由于方法验证的目的和内容决定的,但如果生产工艺或处方的改变、质量标准限度或分析方法发生变化包括稀释剂的改变,都应该进行方法再验证,方法再验证不代表全验证,验证哪些项目依具体情况进行评估后确定,可能评估结果是再验证只需进行专属性验证,甚至可能不需要进行再验证。
参考文献:
[1] 国家药典委员会.中华人民共和国药典(2020 版)四部通则增修订征求意见稿.9101分析方法验证指导原则[S].
[2]U.S. Pharmacopoeia 38[M].<1225> Validation of Compendial Procedures.2015:1445-52.
[3]TheInternational Conference on Harmonisatiaon of techniacal Requirements forRegistration of Pharmaceuticals for Human Use (ICH).Q2 (R1) Validation ofAnalytical Procedures: Text and Mothodology[S].2005.
[4]MariaKristina Parr,Alexander H.Schmidt. Life cycle management of analyticalmethods[J].Journal of Pharmaceuticak and Biomedical Analysis,2018,147:506-517.
[5] 国家药典委员会.中华人民共和国药典(2020 版)四部通则增修订征求意见稿.0512高效液相色谱法[S].
[6] U.S. Pharmacopoeia 39[M].<621> Chromatography2016: 464-465.
[7] European Pharmacopoeia9.2.Chromatographic separation techniques:4289.
[8]余盛刚,魏远龙等.一个新的色谱分离度公式[J].齐齐哈尔大学学报,2007,23(4):1-4.
[9]《化学药物杂质研究的技术指导原则》课题研究组.化学药物杂质研究的技术指导原则[S].2015:7.
[10] 国家药典委员会.中华人民共和国药典(2020 版)四部通则增修订征求意见稿.新增分析方法确认指导原则[S].
[11] 国家药典委员会.中华人民共和国药典(2020 版)四部通则增修订征求意见稿.新增分析方法转移指导原则[S].

来源:药事纵横


