您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-05-20 09:43
引言
⊙口服固体制剂生产中的物料为何需要制定粒度标准?
⊙粉末直压、湿法、干法、微丸生产的过程产品粒度分布如何考虑?
⊙粒度标准如何建立才能达到控制产品质量的一致性?
⊙粒度方法学验证与常规方法验证的区别是什么?
⊙如何科学制定粒度的可接受标准?
带着上述疑问,让我们一同走进由多位FDA审评专家撰写的文章Particle Size Specifications for Solid Oral Dosage Forms: A Regulatory Perspective,本文将给出全面的解析,值得一读!值得收藏!
1、前言
在制药行业中,粉体的颗粒特性已成为口服固体制剂产品开发和质量控制中至关重要的因素之一。原料药的粒度分布(Particle Size Distribution,PSD),可能会对终产品的性能产生显著的影响(如:溶解度、生物利用度、含量均匀度、稳定性等)。此外,原料药和辅料的粒度分布也会影响药物的可生产性(如:流动性、总混均匀度、可压性等),最终可能影响药物的安全性、有效性和质量。很多出版物中都提到,粉体的粒度分布对口服固体制剂生产过程中的每一步都有很大影响,包括预混合/混合、制粒、干燥、整粒、包衣、包装和压片[1-3]。因此,在每个特定药物申报的不同开发阶段,应评估药物生产过程中粉体的粒度影响[4]。一旦在最终开发阶段确定了粒度的影响,就可以选择粉体的粒度分布,并确定合适的质量标准,达到控制产品质量的目的,保证生产的一致性。
本文将从监管的角度,对口服固体制剂粒度标准制定的几个重要方面进行讨论。讨论将包括粒度作为处方组成和中间产品关键物料属性的一部分,在建立粒度标准方面的关键考虑。
2、何时需要制定粒度标准?
对于原料药,ICHQ6A指导原则中提出了何时需要制定粒度标准的建议(决策树#3)[5]。总之,如果原料药的粒度对药品的性能(如:溶出度、溶解度、生物利用度、含量均匀度、稳定性或产品外观)或产品的可生产性(即工艺可行性)至关重要,那么就需要建立一个粒度标准。然而,在许多新药申请(NDAs)和仿制药申请(ANDAs)中,对粒度的控制通常被认为是对药品性能的控制,而对产品的可生产性(如:流动性、混合均匀度、可压性等)的影响通常未被考虑。例如,在粒度标准建立时,只关注了低溶解性原料药(对生物利用度有影响)或低剂量药物(对含量均匀度的关注)。如果药品不是低剂量药物,同时原料药是高溶解性的,那么粒度标准就可能不用建立或制定时不用考虑那么多或根本不用考虑其科学性。
对于口服固体制剂,粒度对产品的可生产性的影响是非常显著的,因为其生产过程中几乎每一个工序与粒度相关。例如,制粒和包衣会与粒度增大有关,而粉碎和研磨(milling and grinding)与粒度减小有关。过筛和筛分(screening and sieving)会与不同粒度的颗粒分离有关。混合和混匀(mixing and blending)与不同组分的粒子混合有关,这些组分间的粒度差异对混合均匀性都有很大影响。因此,在每个药物的申请中,理解粉体粒度对生产工艺的影响至关重要。对于原料或过程中的物料(in-process material),如果它们的粒度对生产工艺(如:混合、制粒、整粒、总混、包衣等)影响较大,那么有必要对这些粉体进行粒度控制,以确保生产的一致性。因此,每种物料都需要制定粒度标准,不仅包括原料药、辅料也包括过程中的物料。接下来将讨论几个生产工艺,以证明何时需要建立口服固体制剂的粒度标准。
2.1 片剂生产:直接压片法
直接压片是指原料药和合适的辅料(包括填充剂、崩解剂和润滑剂等)混合均匀后在模具内直接压制成片剂的过程[6]。对于这些不同组分(如原料药、填充剂、崩解剂、润滑剂等),如果颗粒大小、形状或密度存在显著差异,混合粉(如混合物)可能有分层的趋势,这将导致混合均匀度差。分层可能主要是出现在大小差异的组分中,性状、密度上有显著的差异,可能会造成在混合物中发生分离,此时,颗粒形态和密度的差异则是次要因素[7]。此外,由于原料药和辅料颗粒仍存在于混合物中,每个成分都以独立的颗粒形态呈现出来。尽管最终的混合物符合混合均匀性的接受标准,但由于混合后不同组分的颗粒大小、形状或密度不同,原料药可能会在混合后出现分离或结块。因此,为了确保生产过程的一致性,原料药和关键辅料都需制定粒度标准。另外,如果颗粒形态对最终混合物粉末的混合和流动性均有很大影响,则需要提供颗粒形态的信息。在以往文献中,有报道过颗粒形态对混合和压片过程的影响[8-9]。
2.2 片剂生产:制粒
除直接压片外,片剂还可以通过湿法或干法制粒方式来生产。制粒本质上是颗粒放大过程的一种,既能提高流动性,又能提高压片特性[6]。在干法制粒过程中,原料药和稀释剂的粉体颗粒会在高压下聚集(通过挤压形成带状形式),然后在压制前进行粉碎和筛分。在湿法制粒过程中,将溶液加入到原料药和稀释剂的粉末中产生聚结,然后在压片前,进行干燥、整粒和过筛制成颗粒。在这两种情况下,由于单一原料药和稀释剂在制粒过程中被颗粒化(至少在一定程度上),所以片剂的质量直接受到颗粒(原料药和稀释剂的混合物)的影响,而不是单个原料药或稀释剂颗粒的影响。因此,控制粒度分布和颗粒流动性对最终的混合和压片过程具有潜在的关键作用。
正如美国FDA口服固体制剂研发和验证批准前及批准后检查指南(1994)中所指出的那样:在制粒中,用于说明批间粒度一致性的主要物理参数是颗粒的粒度。这对生物批(biobatch)与生产批进行比较以及对工艺修改或变更同样重要。粒度会提供有用的可比性信息。颗粒的大小,甚至是颗粒的类型都会影响片的孔隙大小,并对溶出产生影响。例如,包衣片的溶出失败可能是由于整粒筛网大小变化引起的,产生了较大的颗粒,导致很低的溶出[10]。因此,颗粒的粒度标准建议作为制粒工序的一部分。由于在湿法或干法制粒过程中有一个预混合过程,如果这些成分对总混粉末或成品的均匀性有显著影响,那么就需要对原料药和关键辅料的粒度分布进行控制。
2.3 多层包衣缓释胶囊的生产
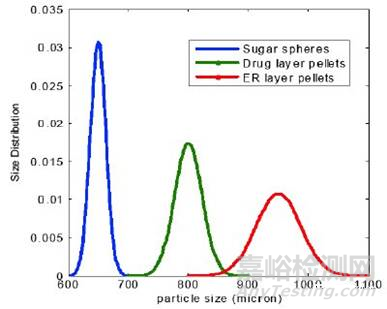
许多缓释制剂(ER)都是由小的包衣糖丸填充到胶囊中制成的,这些小丸的生产涉及到药物层包衣和缓释层包衣过程,本质上是颗粒增大的过程。在多层包衣工艺过程中,颗粒大小的增加如图1所示。在不同包衣阶段对颗粒大小的变化进行监测和控制,可为控制产品的质量提供有效方法。Heinicket等人研究了颗粒大小与包衣厚度之间的关系[11],并证明了聚合物包衣厚度可以通过动态成像分析(DynamicImaging Analysis,DIA)来测量,这为多颗粒产品的包衣提供了一种潜在的替代分解测试方法[12]。除了成像分析,还可以用简单的筛分分析方法对不同阶段的颗粒大小的变化进行定量评估。
图1-多层包衣过程中小丸粒度分布的变化
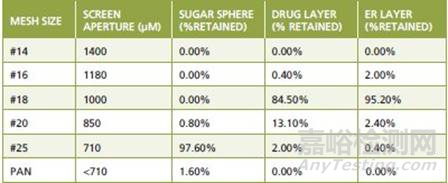
表1中显示了在不同的生产阶段如何改变颗粒大小分布的一个例子。对于批间一致性,这些颗粒粒径的测定在不同阶段都提供了有用的信息。此外,颗粒宽的粒度分布可能会导致颗粒的分离,并会影响到颗粒的均匀性,因此,颗粒大小分布的控制不仅对包衣有重要的影响,而且对混合过程也很重要。在此,对初始的糖球、包药物层颗粒、包缓释层颗粒和最终混合颗粒,建议制定可接受的粒度标准。针对原料药,如果其完全溶解在包衣溶液中,一般不需要制定粒度标准。但是,如果原料药混悬在包衣液中,那么也最好制定一个粒度标准。
表1-多层包衣过程中小丸粒度分布的改变
此外,除了控制粒度分布外,药物层和缓释层的包衣均匀性是产品质量的关键因素。由于小丸的体积小,重量的测定对包衣均匀性的控制是没有意义的。相反,对于每个包衣阶段,包衣均匀性应在适当的取样计划下,通过收集到的多个位置的单位剂量样品中原料药含量和含量均匀度来评估的。当原料药平均含量与预期显著不同或不同位置间存在较大差异时,包衣过程可能未得到很好的控制。同样,在封装前的终混器或容器中,通过对多个位置采取合适的取样方式所收集到的样品进行溶出度测试,是评价缓释层包衣均匀性的一个有用方法。
3、如何建立合适的粒度标准?
一旦确定需要制定粉体的粒度标准,下一个问题就是如何建立一个恰当的粒度标准。许多新药申报和仿制药申报资料中,由于粒度标准在制定时考虑不充分,无法达到控制粉体粒度分布的要求。因此,为了满足法规的要求,了解粒度标准中应包含哪些信息就显得尤为重要。在ICH Q6A指导原则中,质量标准以测试列表的形式被定义出来,并对参考方法、适合的接受标准以及测试数值的限度、范围或其他要求做了描述。接下去我们将从一个合适的粒度标准建立过程展开讨论,其中包括分析方法、方法验证和可接受标准。筛分法和激光衍射法是口服固体制剂最常用的粒度测定方法,以下将围绕这两个方法开展探讨。
3.1分析方法
关于分析方法,USP通则<786>和<429>中分别提供了筛分法和激光衍射法的具体要求。但是,在申报资料中,很多并没有完全遵循USP的要求。
一般来说,筛分法或激光衍射法测定粒度包括以下步骤:(1)粉末的抽样,(2)对抽样样品进行取样作为分析样品,(3)样品的制备或分散,(4)仪器设置和确认,(6)粒径测定,(6)数据分析和说明,(7)报告粒度结果[13]。因此,一个完整的分析方法应该包括所有这些信息。在这些阶段中,如何从粉体中获得少量能代表粒度分布的样品很关键。Allen已经证明,选择合适的取样器将大大提高粒度测定的重现性[14]。另外,对于粒径小或有粘性的颗粒,选择合适的样品分散方法也是至关重要的,这些颗粒有聚集的趋势。样品分散的目的是尽可能地减弱样品分析中颗粒的聚集,同时避免过度使用分散力而造成颗粒损耗。通常来说,在粒度测定中出现的误差大多是由于取样或样品分散缺陷造成,而不是因为仪器问题产生的。因此,粒度标准中,建议在分析方法部分详细描述取样和样品分散方面的具体操作。
对于激光衍射法,数据分析和说明同样重要,若光学模型选择不合适(如Mie或Runover理论)或折射率值都有可能会导致粒度分布的显著偏差[15]。因此,为了得到更好的重现性,有必要在报告中将光学模型和折射率值写清楚。硬件和软件方面同样存在一些明显差异,不仅是不同厂商的设备,甚至连同一厂商的不同型号设备都存在很大差别,因此,建议提供激光衍射仪的足够信息,包括用于分析使用的软件。
3.2分析方法验证
分析方法验证是通过实验室研究来证明方法的性能参数符合预期的分析应用目的的过程[16]。由于粒度分析方法的独特性,其方法验证不同于ICH Q2A、Q2B以及USP通则<1225>[17-18]中描述的验证内容。一般来说,粒度方法验证通常需要对精密度(重复性和中间精密度)和耐用性进行评价。在ICH中涉及到的其他验证参数,如专属性、线性与范围、准确度、检测限和定量限等都不作为粒度方法验证的要求。
精密度中的重复性是指在短时间内,由实验室的同一实验人员使用同一台仪器进行验证的过程。中间精密度是指在同一实验室,在不同时间由不同实验人员使用不同仪器进行测定的过程[16]。如果将粒度方法转移到其他实验室,则需要对不同实验室所使用的方法进行重现性评价。
分析方法的耐用性是指测量条件有小的变化时测定结果不受影响,对方法参数进行故意改变并提供正常使用过程中的可靠性指标[16]。在粒度测定方法的开发阶段,应对耐用性进行评估。一旦确定了最优的方法参数,就可以通过这些参数的微小变化来评价耐用性。对于筛分法,推荐以下参数来验证耐用性:(1)样本量,(2)拍击速率,(3)旋转速率,(4)搅拌时间。对于激光衍射法,推荐下列参数来验证耐用性:(1)测量稳定性,(2)折射率,(3)分散压力(干法测定),(4)样品浓度,超声和搅拌速率(湿法测定)。耐用性应该在方法参数故意发生变化时显示出分析的可靠性。在分析方法验证报告中,也应包括能表明在分析过程中所描述的取样和样本分散策略的可靠性和重复性的研究。此外,建议使用图像分析作为评估颗粒形状、大小范围以及选择的筛分法或激光衍射法适用性的辅助工具。
3.3可接受标准
可接受标准是指对分析方法结果的限度、范围或其他参数的衡量标准[5]。对于粒度标准,可接受范围应与产品性能或可生产性联系起来。通常需要的是一个窄的粉体粒度分布。对于相对较广的粒度分布,控制整体粒度分布比只对平均粒径控制要重要得多。两种平均粒径相同但粒径分布不同的颗粒,其对产品性能或可生产性的影响可能会非常大。
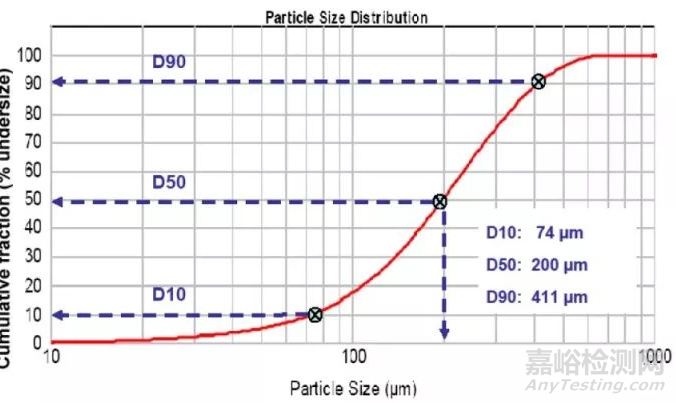
对于激光衍射法,D10、D50和D90的限度常被作为可接受标准。这些参数代表的是样品中一定大小颗粒的累积分布百分比,D10、D50或D90指的是累积粒度分布为10%、50%和90%的对应值,其代表了粒径小于它的比例占10%、50%和90%。例如,图2中通过激光衍射法测定得到的粒径大小数据被绘制成累积粒度分布。它表明D50是200μm,代表50%的粒径低于200μm(即:中值粒径)。类似的,D10和D90分别是74μm和411μm,这表明10%的粒径低于74μm,90%的粒径都低于411μm。应该注意的是,对于单一粒子的测定,D10、D50和D90不是一个范围,而是一个值。这些值在不同的测定或不同的样品中会有所改变。
图2-激光衍射法测定累计粒度分布结果
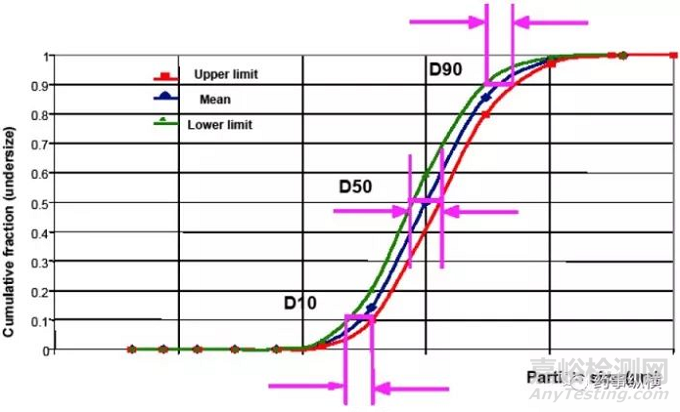
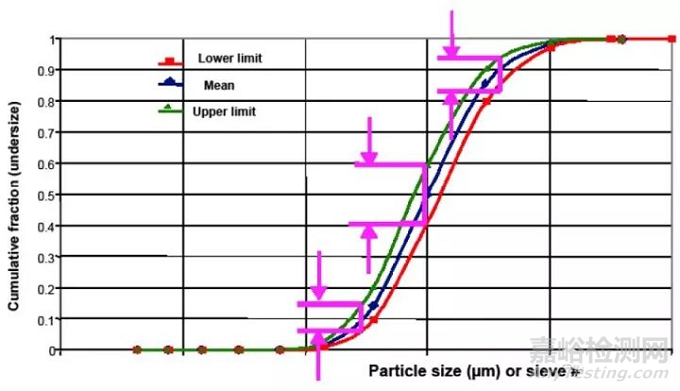
粒度分布可以通过D10、D50和D90分别设置上限和下限来控制。如图3所示,如果蓝线上的钻石点表示目标或期望的粒度分布,D10、D50和D90的上限和下限提供了样品粒度分布变化的控制策略,使得绿线和红线之间的样品没有对产品性能或生产造成不利影响。设置上限和下限值,很大程度上取决于粒度分布的影响有多大,这通常是基于先前的理论或实验设计(DOE)的结果。如何设置这些限度,需要一定的支持性数据来证明,同时也应该包括在药品申报中。在许多NDA和ANDAs中,仅对D10、D50或D90建立了上限或下限。在没有科学依据的情况下,这种单侧限度是不可接受的,因为它不能对粉体的粒度分布进行充分的控制。
图3-采用激光衍射法测定的粒度分布的D10、D50和D90的接受标准。蓝线带有钻石点的蓝线表示目标或期望的粒度部分。三角形点的绿线代表了粒度分布的下限。带点的红线代表了粒度分布的上限。
对于筛分法,建议测量样品的整个粒径分布,而不是测定一两个筛子上的颗粒比例。一旦从筛分法测定中得到累积粒度分布,就可以通过激光衍射法中使用的类似方法来确定D10、D50和D90的接受标准。如图4所示,更常见的情况是三个筛号(或粒度)的累积比例的上限和下限被定为可接受标准。在这种情况下,不是控制大小变化,而是控制三个固定筛分的累积比例的变化,以确保更好的控制粒度分布。因此,建议选择三个筛子,目标累积量约为10%、50%和90%,以充分代表整个粒度分布。
图4-采用筛分法测定的粒子粒度分布的接受标准。带有钻石点的蓝线表示目标或期望的粒度分布。带有三角形点的绿线代表了粒度分布的上限。用方点表示的红线表示粒度分布的下限。
4、结 论
本文从法规的角度,对口服固体制剂的粒度标准进行了探讨。口服固体制剂的生产与粉体粒度息息相关,粒度是一个重要的影响因素。除了原料药和辅料需要建立粒度标准外,过程产品必要时也需制定标准,以保证产品质量的一致性。
在注册申报中,建议粒度标准应提供完整的分析方法,包括取样、样品处理或分散方法、仪器参数设置、数据分析和说明等。粒度分析方法验证报告中应提供重复性、中间精密度和耐用性等验证内容。此外,在报告中最好有取样和样品分散策略方面的研究,以保证方法的可靠性和重现性。对于可接受标准,建议建立上限和下限,以便更好地控制更宽的粒度分布。在资料中性应提交带有支持性数据的限度制定理由。

来源:药事纵横翻译


