您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-04-29 09:53
进入21 世纪,我国新药研究从仿制向创制转轨已成共识。然而,新药创制是系统工程,需要多学科协同作战,难能一蹴而就。但是对我们13 亿人口的大国来说,服药的重要性不亚于吃饱穿暖,是迫在眉睫一天也不能或缺的国计民生大事。根据我国的实际情况,新药研究应以开发那些结构类型已知,疗效优于或近于现有同类产品的药物作为主攻方向。前药原理是将已知有生物活性而又存在某些缺点(如: 生物利用度差、性质不稳定、作用时间短、有异味等)的药物经结构修饰制成新药即前药,后者体外无活性,在体内分解释放出原药产生药效。与原药相比,前药保持或增强原药的药效,又克服原药的缺点。前药属于结构类型已知,疗效优于或近于现有同类药物的创新药物类型,其特点为投资少,风险小,成功率高,因而在新药研究中占有重要地位,尤其适合目前我国制药工业中既有的实际情况。为推动我国新药研究工作的发展,现按照结构修饰类型综述有关前药原理在新药设计中的应用。
1含羧基药物的前药设计
1.1 成酯前药设计
氨苄青霉素是耐酸、广谱、半合成青霉素,可以口服,但是口服吸收差,血药浓度只有注射给药的20%-40%。分析结构表明,氨苄青霉素分子中C2羧基与C6侧链氨基,在胃内pH情况下解离为两性离子,极性大是影响口服吸收的关键。将羧基成酯,发现其简单的脂肪或芳香酯类不够活泼,在体内酶促分解成原药的速度很慢,血药浓度达不到峰值,其原因是氨苄青霉素分子中羧基邻位的两个甲基占有较大空间,其屏蔽作用阻碍酯酶水解所致。而将其设计成双酯型前药,末端酯键位阻较小,易于发生酶促断裂,生成的羟甲酯不稳定,自动分解,释放出甲醛和氨苄青霉素,产生药效,生物利用度提高3-5倍,口服几乎定量吸收(98%-99%)。
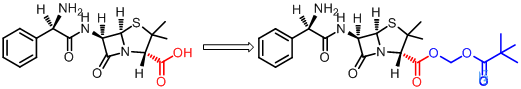
近几年,这种双酯前药设计广泛应用于含羧基药物的前药设计中。
1.2 成醛前药设计
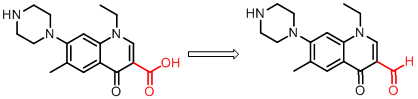
含羧基药物制成醛基前药,可增加原药的脂溶性,显著提高口服吸收效果,增加血药浓度。如氟哌酸,为广谱抗菌药,作用强但口服吸收不完全,只有给药剂量的35%~40%,其原因为分子中羧基与哌嗪环上的氮原子成两性离子,不易透过生物膜,做成酯不理想,做成醛以后,在体内经氧化形成酸,口服吸收好,血药浓度高,因而含羧酸药物成酯不理想时,可考虑做成醛化物一试。
2含羟基药物的前药设计
2.1氨基酸酯前药设计
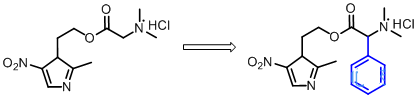
氨基酸的羧基与母药的羟基成酯,其氨基与无机酸成盐,以增加药物水溶性。如甲硝唑-N,N-二甲基甘氨酸酯盐酸盐,水溶性好,血浆浓度高,但水溶液不稳定,需在临用前配制。其原因为分子中的氨基在制剂pH 值为3-5下质子化,有强的吸电子效应,活化了酯羰基,易受OH-离子进攻,使酯键断裂。研究发现,若在酯基和氨基之间引入一个苯基,成为N-取代的胺甲基苯甲酸酯,可完全阻止氨基对酯键的影响,又不影响体内酶促水解反应,如甲硝唑的这种前药水溶性比母药有所增强,水溶液稳定性增加,同样条件下可保存14 年。
2.2磷酸酯前药设计
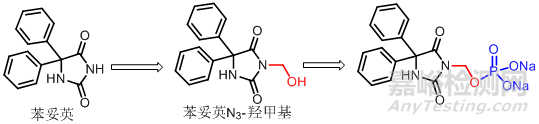
磷酸为三元酸,其单酯钠盐水溶液稳定,体内分解速度快,血浓度高,常用于水溶性前药的制备。如苯妥英N3-羟甲基衍生物与磷酸成单酯钠盐,水溶度增加4000倍,化学稳定性好,便于储存,在体内经酯酶水解生成N3-羟甲基化合物不稳定,迅速脱甲醛产生苯妥英发挥药效。
3含羰基药物的前药设计
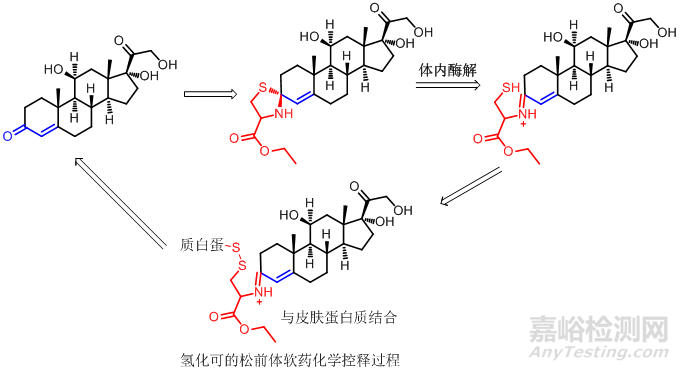
分子中含羰基的药物很多,目前引人注目的前药有希夫氏碱、肟、四氢噻唑、四氢恶唑烯醇酯。如氢化可的松是内源性糖皮质激素,局部用药时可使体内浓度高于正常水平而引起一系列全身副反应,研究表明,分子中α,β-不饱合酮的3-酮基是活性主要部分,因而将其制成前体药物,选用半胱氨酸酯做前体基团制备了酮基前药。局部应用时,该前药比母体药物活性大,全身毒性小。其机制可能是在体内噻唑环自发开环断裂C-S 键,生成希夫氏碱中间体,再与皮肤内细胞的巯基结合而积蓄于局部皮肤,在炎症部位慢慢水解释放出母体药物,从而减少全身毒副作用。螺噻唑烷前药的方法,已用于有抗癌活性的α,β-不饱合醛酮类药物以延长作用时间和降低毒性。
4开链药物的环状前药设计
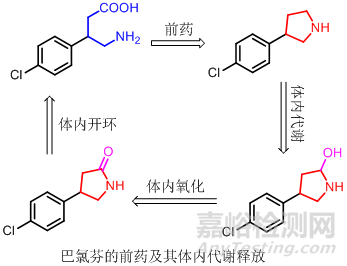
开链药物分子中含有两个或两个以上可衍化的基团,通过适当方法把这些基团桥连起来,生成环状前药,以改善药物原有的理化性质,产生理想的药物疗效。如巴氯芬为γ-氨基丁酸的类似物,有防治癫痫的作用。但由于其高度离子性,给药后进入中枢神经系统的药物不到1%,将其制成环状化合物3-(对氯苯基)-四氢吡咯,在体内经羟基化开环,再经氧化形成巴氯芬产生药效。由于四氢吡咯化合物为非极性分子,易于进入中枢神经系统而药效增加。
前药原理在新药设计中广泛应用,不仅能增加原药的溶解性,以改变药物的给药途径;改善药物的生物利用度,增强药效;还可以矫正药物苦味,异味,使之易于服用,另外还可增加药物的代谢稳定性,以延长药物作用时间等等。总之,对于药剂学不能改善的那些药物的不利因素均可用前药的方法加以纠正,因而,前药原理是新药设计的一条重要途径。
5环状药物的开环前药设计
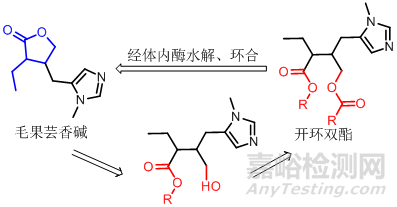
某些环状药物如果在环上引入某些前药基团有困难时,可考虑制成各种开环衍生物作为前药,改善其脂溶性和水溶性。如毛果芸香碱是治疗青光眼的常用药物,滴眼时不易渗入角膜,生物利用度低,作用时间短,需频繁给药。将其制成开环双酯衍生物稳定性增加,室温可贮存5年以上,生物利用度提高,作用时间延长,在体内经酯酶水解、环合,能定量形成毛果芸香碱。
6氨基酰胺类药物的前药设计
6.1酰胺前药设计
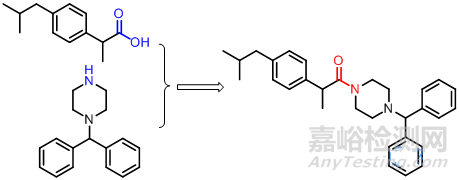
布洛芬为芳基烷酸类非甾体抗炎药,具有较强的抗炎镇痛作用,它不仅能抑制前列腺素的合成,而且对组胺、羟色胺、激肽及补体等其他炎症介质都有不同程度的抑制作用。哌嗪类抗组织胺药作用强而持久,较少产生嗜睡,属于强效、长效H1受体阻断剂,二苯甲基哌嗪是其抗变态反应的活性必要部分,将布洛芬与二苯甲基哌嗪拼合成酰胺产生协同作用,既增强抗变态反应的作用又有较强的抗炎活性,同时减少羧基对胃肠道的直接刺激。
6.2 偶氮化合物前药设计
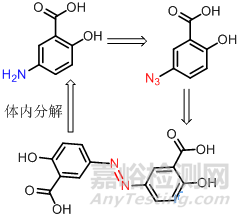
5-氨基水杨酸是治疗溃疡性结肠炎的有效药物,但口服后通过胃肠道时大部分剂量已被吸收,因而在结肠不能达到有效浓度,将5-氨基水杨酸重氮化后,与水杨酸偶合成偶氮水杨酸,口服后几乎不被小肠吸收或分解,到达结肠后才被分解成两分子活性的5-氨基水杨酸,而无其他不良反应,疗效显著。
该前药的分解是利用细菌体内的酶进行催化分解的。
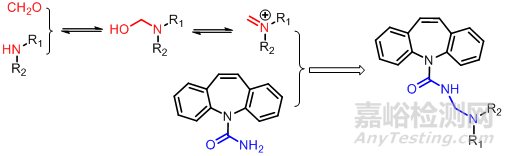
6.3曼尼希碱前药设计
许多酰胺、亚胺、内酰胺及其他酸性类似物与适当的醛、胺反应,得到相应的曼尼希碱可获得理化性质较好的前药,如: 抗癫痫药酰胺咪嗪,水中几乎不溶,只能口服给药,将其与甲醛和二乙胺、二丙胺、哌啶反应形成不同的曼尼希碱,再做成盐酸盐,水溶性比母药大10倍,血浆浓度比酰胺咪嗪高,显效迅速疗效好。
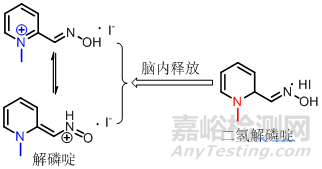
7季铵型药物的前药设计
解磷啶为季铵型有机磷中毒的解毒剂,极性大不易进入中枢神经系统,将其氢化为二氢解磷啶,为叔胺型药物容易透过血脑屏障,进入中枢神经系统,在脑内迅速转化为解磷啶,脑内浓度比服用解磷啶高13倍,本方法成为改善季铵盐类药物在全身及特定部位吸收的有效方法。
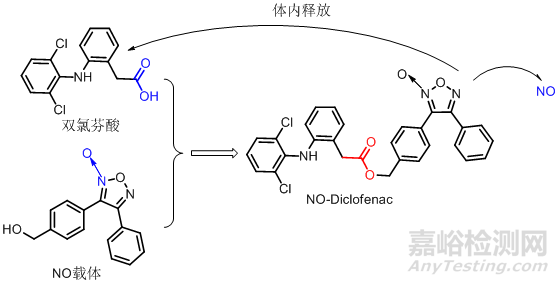
8一氧化氮载体型前药
非甾体抗炎药(NSAIDS) 是治疗风湿性、类风湿性关节炎和骨关节炎的重要药物,疗效肯定但长期用药易引起胃肠道溃疡、出血、穿孔等,因此患者很难坚持长期用药,这是由于NSAIDS抑制了对胃肠道有保护作用的PGS,使肠道中的黏液、碳酸盐分泌减少,黏膜血流量下降,白细胞向血管内皮处大量聚集而产生的。近来药理学研究表明:NO是一种信使物质,参与体内多种生理功能的调节,能抑制胃酸分泌促进黏液分泌,调节黏膜血流量,抑制白细胞的黏附和激活调节生长因子,直接促进溃疡愈合。设想在非甾体抗炎药结构上偶联一个能产生NO的部分,当药物进入体内后,立即释放NO和NSAIDS,NSAIDS可在体内通过抑制COX的活性继续发挥作用,而NO则供给胃肠道的内皮,通过抑制嗜中性细胞聚集,增加黏膜血流量和黏液分泌,以及减少自由基生成等四个方面的作用,减少胃肠道副作用如:NO-Aspirin和NO-Diclofenac具有良好的抗炎抗血栓作用,但对胃肠道损伤明显减少NO-KetoProfen和 NO-Indobufen 等抗炎活性都高于母体药物,且无COX-2心脏病风险,是一类有前途的非甾体抗炎药的替代药。
前药原理在新药设计中广泛应用,不仅能增加原药的溶解性,以改变药物的给药途径;改善药物的生物利用度,增强药效;还可以矫正药物苦味,异味,使之易于服用,另外还可增加药物的代谢稳定性,以延长药物作用时间等等。总之,对于药剂学不能改善的那些药物的不利因素均可用前药的方法加以纠正,因而,前药原理是新药设计的一条重要途径。
〔摘自程辉.中药饮片企业监管思考和探析[J].中国食品药品监管.2019.3(182):75-78.原文有删减〕

来源:药渡


