您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2018-05-02 09:37
编者:溶出度检查方法的分辨力是溶出度检查方法研究的重要内容,选择具有良好分辨力的溶出度检查方法是药品技术审评中关注的内容。本文参考相关文献,介绍溶出度检查方法分辨力的意义和一般方法,并以非布司他片为例探讨建立具有分辨力的溶出度检查方法。
溶出度系指药物从片剂、胶囊剂和颗粒剂等固体制剂在规定的条件下溶出的速率和程度。它是一种模拟口服固体制剂在胃肠道中的崩解和溶出的体外试验法,是药品生产和质量控制的一项重要内容,对保证制剂批间质量的一致性有重要作用。同时,当药品处方、生产工艺、生产地点和生产规模等发生变更后,溶出度检查是比较变更前后产品相似性或差异程度的重要方法和研究工作的重要内容。
通过对试验仪器、溶出介质、转速等的研究建立有效的溶出度试验条件,是固体制剂质量控制研究的重要内容。溶出度检查方法的分辨力是溶出度检查方法研究的重要内容和选择溶出度检查方法的重要依据,也是技术审评中重点关注的内容。
1 溶出度检查方法的分辨力
溶出度检查方法的分辨力是指方法可以发现和区分制剂质量变化的能力。一个有效的溶出度检查方法应当能够发现可能影响制剂的生物药剂学行为的处方、生产工艺等方面的变化。
可能影响药品质量和生物利用度的因素包括原料药供货来源、粒度和晶型,辅料的型号、级别和处方中辅料的用量,以及生产工艺等。例如,对于水难溶药物,不同批次的原料药如果在粒度方面存在差异,不但可能影响药品的体外溶出行为,甚至可能影响药品的生物利用度。为有效保证和控制药品批间质量的一致性,溶出度检查方法学研究中需要注意对方法的分辨力的考察。
2 溶出度检查方法分辨力的基本研究方法
首先需要研究和了解原料药在不同pH值条件下的溶解度,或在不同溶剂中的溶解度,并注意考察药物在不同pH值条件下和不同溶剂中的稳定性。
溶出度试验条件(方法、介质、转速)的选择可参照药品相关技术要求和专业文献进行。对溶出度检查方法分辨力的考察应基于可能影响药品质量和生物药剂学行为的因素的分析,进行相应的研究。
对于水溶解性差的原料药,其粒度、晶型可能是影响制剂溶出行为。可以选择不同粒度、不同晶型的原料药制成制剂,考察这些制剂的溶出行为的差异。
可以根据制剂特性和处方和生产工艺中可能影响药品质量和生物利用度的关键因素,设计一些“异常状态”生产的制剂,考察溶出度检查方法的分辨力。例如,水难溶性药物口服固体制剂处方中最有可能影响药品质量的辅料可能是崩解剂,可以考虑降低崩解剂用量或删除崩解剂袁考察这些制剂的溶出行为的差异。
3 研究实例—非布司他片
3.1 非布司他相关理化性质
非布司他化学结构式见图1:
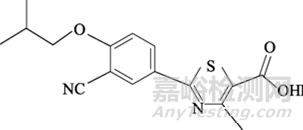
图1 非布司他化学结构式
非布司他的pKa为3.3
非布司他原料药易溶于二甲基甲酰胺,溶于二甲基亚砜,略溶于乙醇,微溶于甲醇或乙腈,几乎不溶于水或酸性溶液。
日本帝人株式会社在中国申请专利(公开号CN1275126A)的说明书和实施例中,明确发现非布司他存在6种晶型,不同晶型的表观溶解度和饱和溶解度无显著差异。根据不同晶型特性,工业化生产优选晶型A。
非布司他(晶型A)在不同溶剂(20±5)℃中的溶解度见表1。
非布司他溶解度随pH值升高而增大,非布司他(晶型A)在不同pH值的缓冲液中(20±5)℃中的溶解度见表2。
3.2 非布司他片的溶出度检查方法


非布司他片2009年2月13日获得FDA批准在美国上市,规格40mg、80mg,FDA公布的非布司他片溶出度检查方法为:浆法,以0.05mol/L磷酸钾缓冲液(pH6.8±0.05)900ml为溶出介质,转速为(65±3)r/min,规定30min溶出大于80%。
3.3 非布司他片溶出度检查方法
国内申报的非布司他片规格为40mg和80mg,非布司他几乎不溶于水和酸性溶液,研究显示采用桨法,转速为50r/min,溶出介质体积900ml,非布司他片在0.1mol/L盐酸溶液中60min溶出低于5%,在pH值醋酸盐缓冲液60min,溶出低于10%,在水中60min溶出低于50%,均不能完全溶出。
非布司他溶解度随pH值升高而增大,在pH6.8磷酸盐缓冲液中37℃溶解度约1.4mg/ml,满足漏槽条件,采用上述溶出度检查条件,以pH6.8磷酸盐缓冲液900ml为溶出介质,非布司他片迅速溶出,10min溶出90%以上。
非布司他几乎不溶于水,参考日本帝人株式会社申请的中国专利CN1642546的权利要求和实施例4的原料药粒度情况,将非布司他原料药经微粉化处理,得到≤25um、≤50um、≤75um、≤100um4个粒径范围的原料药样品,制成片剂后进行溶出行为考察。结果显示,以pH6.8磷酸盐缓冲液为溶出介质,4种不同粒径原料药制备的非布司他片溶出行为相似,,3min均溶出90%以上,不能反映产品的内在质量。
拟在新版美国药典中增加的《1092》章节“溶出度检查方法的建立和验证”中明确指出:溶出介质的选择是根据药物溶解度和制剂规格确定的,以保证符合漏槽条件(定义为至少3倍于药物饱和浓度体积的介质体积)。为得到可靠的溶出度数据,可以考虑加入表面活性剂。选择添加不同量(0.1%~0.5%)十二烷基硫酸钠水溶液作为非布司他片溶出介质,结果显示,4种不同粒径原料制备的非布司他片具有不同的溶出结果,30min分别溶出98%(≤25um)、88%(≤50um)、77%(≤75um)、60%(≤100um),即含适量十二烷基硫酸钠水溶液作为溶出介质可有效地区分不同粒径原料药制备的非布司他片,可以反映产品的内在质量。
研究结果说明,在方法分辨力方面,选择含适量十二烷基硫酸钠水溶液作为溶出介质优于pH6.8磷酸盐缓冲液。
4 结语
一个具有良好分辨力的溶出度检查方法对保证和控制药品批间质量的一致性有重要作用。在进行处方筛选和工艺研究阶段,以及在与原研产品进行溶出行为对比研究时,选择一个具有良好分辨力的溶出度检查方法对研究和技术评价有重要作用。同时,药品获准上市后发生变更时,一个具有良好分辨力的溶出度检查方法对变更前后对比研究具有重要作用。
最理想的溶出度检查方法应可以区分可能影响产品生物利用度的处方、工艺等方面的改变。但是,除非已经明确建立了体内外相关性,溶出行为的改变可能反映制剂体内药代动力学行为的变化,但体外溶出行为的改变也可能与制剂的体内药代动力学行为变化无关,即溶出度检查方法过于敏感,即使体外2种制剂的溶出行为存在差异,但体内是生物等效的,此时,如产品质控无特殊要求,也可以不必采用过于敏感的检查条件[例如对于生物药剂学分类(BCS)I的药品]。因此,在关注溶出度检查方法分辨效力的同时,还需考虑溶出度检查方法的体内外的相关性。

来源:AnyTesting


