您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2025-08-14 18:55
在医疗领域,设备的安全性和有效性不仅取决于其技术性能,更与用户体验息息相关。一个用户交互设计不良的医疗器械,即使功能强大,也可能因操作复杂、界面混乱而导致用户使用错误,甚至危及患者和使用者的生命安全。
欧盟新实施的《医疗器械法规》(MDR, EU 2017/745)取代了原有的MDD指令,对医疗器械的可用性(Usability)提出了更严格的要求,并将其纳入产品的全生命周期管理。
这意味着,制造商不但需要证明设备的技术合规性,还必须保证其整体的设计符合真实用户的需求,从而最大限度地降低使用相关风险。
本文将系统解析MDR对可用性的核心要求,并提供企业应对策略,帮助医疗器械厂商顺利通过合规审核。
01MDR框架下的可用性核心要求
1. 可用性工程(Usability Engineering)成为强制性过程
MDR法规中有明确要求,制造商必须建立系统的可用性工程过程(Usability Engineering Process, UEP),并符合IEC 62366-1标准。该过程涵盖:
用户需求分析(目标用户、使用环境、使用场景)
用户界面设计(硬件、软件、标签、说明书等)
使用风险评估(识别可能的操作错误及其影响)
验证与确认(确保设计满足用户需求)
2. 强调“使用错误”(Use Error)管理
MDR特别关注“使用错误”(即非用户故意导致的错误操作),要求制造商:
预测可能的错误(如按钮误触、界面混淆)
评估错误的影响(是否会导致严重伤害?)
通过设计降低风险(如优化界面布局、增加防错机制)
3. 用户多样性考量
MDR要求设备必须适应不同用户群体,包括:
医护人员(医生、护士、技师等)
患者(老年人、儿童,残障人士、非专业人员等)
使用环境差异(手术室,急诊室、家庭护理、户外急救等)
这意味着,同一款设备可能需要针对不同用户群体进行定制化设计,例如:
家用血糖仪需简化操作,避免复杂步骤
手术设备需考虑医生在高压环境下的操作习惯
02MDR对可用性验证与确认的具体要求
MDR要求制造商进行可用性验证(Verification)和可用性确认(Validation),二者区别如下:
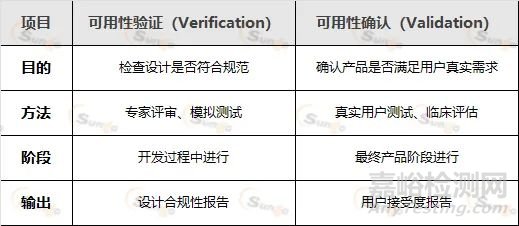
1. 可用性验证(Verification)
专家评审:由人因工程专家评估界面设计是否符合标准
模拟测试:使用原型机进行实验室测试,检查潜在问题
2. 可用性确认(Validation)
用户测试:招募真实用户(如医生、护士)实际操作
临床评估:在真实医疗环境中观察设备使用情况
03如何构建符合MDR的可用性文档?
MDR要求制造商提供完整的可用性工程文档,主要包括:
可用性工程计划(UEP Plan)
描述可用性活动的目标、方法和时间表
用户分析报告(User Analysis)
定义目标用户(如医生、患者)及其能力限制
使用场景分析(Use Scenarios)
描述设备在正常和紧急情况下的使用方式
使用风险评估(Use-Related Risk Analysis)
识别可能的操作错误及其影响
可用性测试报告(Usability Test Report)
包括测试方法、结果和改进措施
关键点:这些文档必须与技术文件(Technical Documentation)和风险管理报告(RMR)中识别的可用性风险, 临床评估报告(CER)保持一致,形成完整的证据链。
04企业如何应对MDR可用性要求?
1. 早期整合可用性工程
在产品概念阶段引入可用性专家避免后期返工
采用迭代设计,不断优化用户体验
2. 跨部门协作
设计团队(优化界面)
工程团队(确保技术可行性)
临床团队(验证真实使用需求)
法规团队(确保符合MDR要求)
3. 真实用户参与测试
邀请医生、护士、患者进行模拟操作
收集用户的反馈并进行相应的优化设计
4. 上市后监督(Post-Market Surveillance)
持续收集真实用户的使用反馈,监测以往可能未识别到的可用性问题,必要时发布设计更新或在说明书中补充有关的安全警示

来源:Internet


