您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2025-07-30 21:31
7月30日,国家药监局审评中心发布《发酵/半合成抗生素仿制药有关物质限度指导原则》,并自发布之日起施行!
该指导原则指出,与化学合成工艺相比,发酵工艺可变性大、可控性低,发酵或半合成原料药的杂质谱通常更复杂且难以预测,因此,ICH Q3A、ICH Q3B 等指导原则的适用范围未涵盖发酵或半合成来源的药物。在限度制定时,需要充分考虑生产工艺的复杂程度、产品特点、药典标准、参比制剂杂质状况等因素。多数情况下,抗生素类药物的治疗持续时间较短,因此,可能接受比ICH Q3A、 ICH Q3B更宽的有关物质限度。
该指导原则规定了发酵或半合成化学仿制药抗生素有关物质的报告限度、鉴定限度和界定限度。如发现存在安全性隐患,应适当收紧限度;如需设定更宽的限度,应进行充分论证。
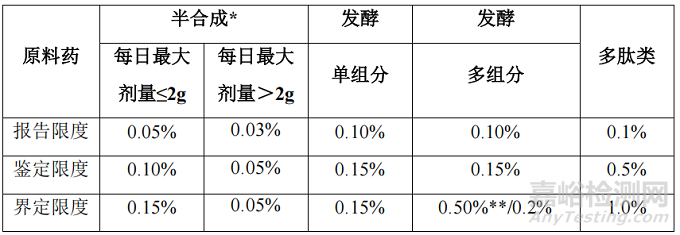
原料药
制剂
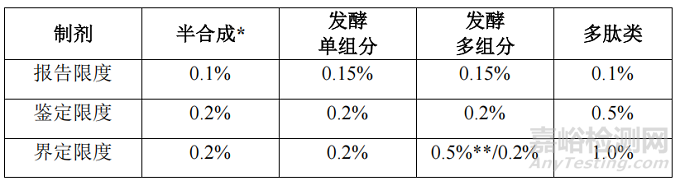
文件分别对半合成原料药、单组分发酵原料药、多组分发酵原料药、多肽类和杂质谱非常复杂的情况,以及制剂的工艺特征、杂质风险进行阐述并给出了杂质限度的建议
文件还对有关物质分析方法给出建议:推荐采用富含杂质的样品(如,杂质加标样品、原料药粗品或分离纯化前样品等)确证方法的专属性。杂质定量方法通常建议采用外标法或加校正因子的主成分自身对照法。对属于弱发色团的抗生素,应筛选适宜的检测方法,确保方法定量限不高于报告限度。对于杂质谱复杂的药物,推荐将分离技术(如,HPLC 法等)与质谱分析等技术联合应用,经充分的合理性论证后,常规检测时可采用较简单的方法。
为进一步明确发酵或半合成化学仿制药抗生素有关物质限度制定研究技术要求,完善化学仿制药抗生素评价标准体系,按照国家药品监督管理局的工作部署,国家药品监督管理局药品评审中心组织制定了《发酵或半合成化学仿制药抗生素有关物质限度制定指导原则》,全文如下。
发酵或半合成化学仿制药抗生素有关物质限度制定指导原则
目 录
一、概述
二、总体考虑
三、原料药的有关物质限度
(一)半合成原料药
(二)单组分发酵原料药
(三)多组分发酵原料药
(四)多肽类
(五)杂质谱非常复杂的情况
四、制剂的有关物质限度
五、分析方法
六、名词解释
七、参考文献
一、概述
抗生素原料药的生产方法主要包括发酵法、半合成法和化学合成法。与化学合成工艺相比,发酵工艺可变性大、可控性低,发酵或半合成原料药的杂质谱通常更复杂且难以预测,因此,ICH Q3A、ICH Q3B 等指导原则的适用范围未涵盖发酵或半合成来源的药物。为明确发酵或半合成化学仿制药抗生素类药物有关物质限度制定的技术要求,在参考国内外相关指导原则的基础上,结合我国化学仿制药抗生素研发和生产现状,制定本指导原则。
本指导原则旨在为发酵或半合成来源的化学仿制药中抗细菌类抗生素原料药及制剂中有关物质研究和限度制定提供一般性指导。发酵或半合成抗真菌化学仿制药可参考本指导原则开展研究。
本指导原则仅代表药品监管部门对于该问题的当前认知,随着相关法规的不断完善以及药物研究技术要求的提高,本指导原则中的相关内容将不断完善与更新。
二、总体考虑
发酵或半合成化学仿制药中抗生素的有关物质研究思路要求与ICH Q3A、ICH Q3B等指导原则基本一致,但在限度制定时,需要充分考虑生产工艺的复杂程度、产品特点、药典标准、参比制剂杂质状况等因素。多数情况下,抗生素类药物的治疗持续时间较短,因此,可能接受比ICH Q3A、 ICH Q3B更宽的有关物质限度。
本指导原则规定了发酵或半合成化学仿制药抗生素有关物质的报告限度、鉴定限度和界定限度。如发现存在安全性隐患,应适当收紧限度;如需设定更宽的限度,应进行充分论证。
本指导原则中的有关物质包括发酵来源的起始物料及其引入的有关物质、发酵或合成副产物、中间体和降解产物等,不涉及发酵工艺中的残留物(如,微生物、培养基、基质和前体的残留物)。
原料药
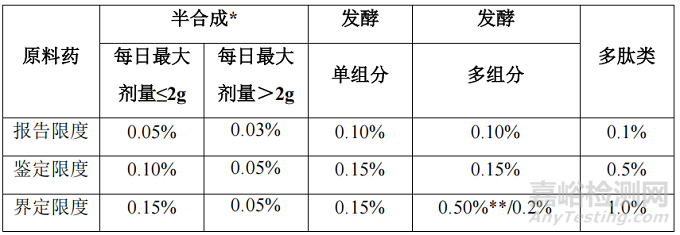
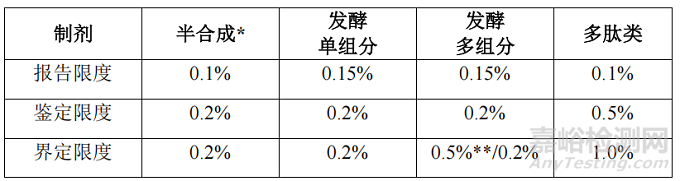
制剂
*)如果原料药属多组分,则可执行“发酵,多组分”的限度。
**)结构密切相关的杂质。
三、原料药的有关物质限度
(一)半合成原料药
多数情况下,半合成工艺中的起始物料或中间体与合成工艺制备的起始物料相似,均是经过良好表征、纯度较高的化合物,因此该类原料药的限度建议参考 ICH Q3A 制定。
如果半合成原料药是由多种活性化合物组成的混合物,经充分的合理性论证,可采用多组分发酵原料药的限度。
(二)单组分发酵原料药
与多组分发酵原料药相比,单组分发酵原料药易于纯化,因此可制定相对严格的限度。
(三)多组分发酵原料药
多组分发酵原料药的组分应明确,并在质量标准中规定各组分的比例,除组分外的化合物建议均按杂质控制。由于多组分发酵原料药组成复杂,通常难以把组分从其他与母体结构密切相关的化合物中纯化出来,且有些结构密切相关的化合物与组分具有相似的抗菌活性。因此,对于结构密切相关的杂质可采用更宽的界定限度,但应提供结构密切相关的证据。
一般情况下,对于半合成或单组分发酵原料药,比提供杂质安全性数据更好的方法是优化生产工艺,使杂质含量满足限度要求。对于多组分发酵原料药,过度纯化可能会导致组分的分布发生变化,应优先保证各生产批次原料药组分的一致性。
(四)多肽类
多肽类抗生素的限度不适用于含有其他修饰部分的肽类(如,糖肽等)。如采用其他限度,应证明其合理性。
(五)杂质谱非常复杂的情况
对于杂质谱非常复杂或者两个杂质极为相似的药物,若现有分析技术无法实现杂质峰的分离,可对未分离的峰设置一个合并的限度,并论证限度的合理性。
对于杂质谱非常复杂且无法对单个杂质峰逐一进行鉴别的药物,建议在与多批参比制剂进行充分对比的前提下,基于足够生产批次样品的杂质谱信息,至少建立一个描述性的质量标准来表征其杂质谱,以确保后续生产批次杂质谱的一致性。
该描述性的质量标准应包括以下参数:
(1)在预设的窄的相对保留时间范围内,规定出现色谱峰的数量及含量总和。
(2)在预设的相对保留时间范围内规定杂质限度(例如:相对保留时间 X 和 Y 之间至少有一个色谱峰,其含量在 A%和 B%之间)。
(3)在预设的相对保留时间范围内,规定超过某一限度值的色谱峰数量(例如:相对保留时间 W 和 Z 之间的任一单个杂质峰含量不得过 C%,但超过 D%的峰不得多于 1 个,其中 C>D,例如,C=2.0%,D=1.5%)。
四、制剂的有关物质限度
除多肽类制剂外,对于剂量较低的发酵或半合成抗生素,如经过合理性论证,可参考 ICH Q3B 指导原则制定鉴定限度和界定限度。
五、分析方法
有关物质分析方法应对潜在杂质具有良好的分离和检出能力,推荐采用富含杂质的样品(如,杂质加标样品、原料药粗品或分离纯化前样品等)确证方法的专属性。杂质定量方法通常建议采用外标法或加校正因子的主成分自身对照法。对属于弱发色团的抗生素,应筛选适宜的检测方法,确保方法定量限不高于报告限度。对于杂质谱复杂的药物,推荐将分离技术(如,HPLC 法等)与质谱分析等技术联合应用,经充分的合理性论证后,常规检测时可采用较简单的方法。
六、名词解释
发酵原料药:本指导原则中,系指自然界存在或通过传统方法或重组 DNA 技术改良的微生物(例如细菌、真菌和微藻等)的代谢产物。
半合成:本指导原则中,系指发酵工艺后进行的一步或多步合成步骤。
单组分:通常系指活性物质仅由一种成份组成。
多组分:系指原料药是由多种活性化合物组成的混合物。
结构密切相关的杂质:系指多组分原料药中,与母体化合物结构密切相关的化合物。判定二者结构密切相关需论证的因素包括:(1)杂质应是已鉴定的杂质;(2)杂质与原料药的组分应具有相近的理化特性(包括光谱特征);(3)杂质与原料药中各组分的主要特征结构应相同;(4)仅允许与原料药中各组分之间结构不同的部分存在差异,典型的差异可能是烷基链的微小变化(不同的支链、少或多一个亚甲基),或不限于烷基链上的氢原子被甲基取代;(5)没有与毒理学高度相关的新的警示结构,比如 N-亚硝基、环氧基以及氧化偶氮基等。
七、参考文献
1. 中国药典 2020 年版四部通则 9102 药品杂质分析指导原则.
2. 国家药品监督管理局. 化学药物杂质研究的技术指导原则. 2005.
3. EMA. Guideline on setting specifications for related impurities in antibiotics. 2012.
4. ICH Q3A(R2): Impurities in new drug substances. 2006.
5. ICH Q3B(R2): Impurities in new drug products. 2006.

来源:Internet


