2025中国药典将于今年10月正式实施,相比2020药典,药品标准进一步完善,药品质量控制和安全保障水平明显提升。企业应紧跟药典的步伐,进一步健全质量管理体系,将质量控制关口前移,强化源头控制。本文结合2025药典要求,对质量管理中有待改进的方面进行了分析汇总。
01 供应商管理
2025药典二部凡例新增元素杂质及残留溶剂要求,规定药品生产企业均应按照相关要求(通则0861、0862)对元素杂质及残留溶剂进行风险评估和控制。
《0861残留溶剂》明确指出:制剂生产企业需要了解原料药和辅料残留溶剂量的相关信息,以符合本原则的规定。
《0862 元素杂质》明确指出:原料药、辅料、包装材料和生产设备供应商提供的关于潜在元素杂质的信息有助于药品生产企业开展元素杂质风险评估。制剂生产企业可以使用原料药或辅料生产企业提供的元素杂质测定数据或者风险评估报告,用于证明最终制剂是否符合本指导原则的限度要求。
从以上要求可以看出,原料药或辅料等生产企业所提供信息,将作为制剂生产企业风险评估的重要信息来源。因此,企业应将“原料药、辅料、包装材料和生产设备供应商提供的残留溶剂和元素杂质的支持性数据/报告”作为供应商资质审核的一部分。
对于残留溶剂的报告方式,《0861残留溶剂》提出了三种建议,企业可视情况选择以下一种作为验收标准:
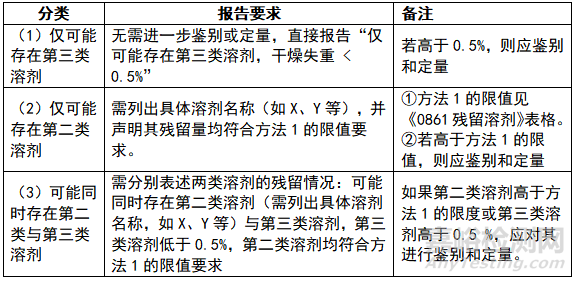
对于元素杂质,企业可以选择“元素杂质测定数据或者风险评估报告”作为验收标准。对于风险评估中建议考察的元素杂质,2025药典按照元素杂质分类、是否是有意添加引入和给药途径列出,企业可参考,详见《0862 元素杂质》表1。
02 试药的管理
试药系指药典中供各项试验用的试剂(不包括各种色谱用的吸附剂、载体与填充剂)。试药的管理通常包括采购、验收、贮存、使用和废弃等。
2.1 提高试药的质量要求
药品检验中使用的试药质量直接影响药品分析检测结果的质量,因此,加强试药的质量管理对于质量控制水平的提高意义重大。
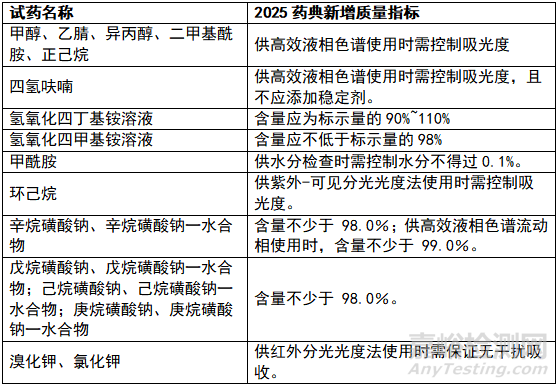
《8001试药》对多种常用试药的关键质量指标提出新的要求,详见下表:
企业应在内部管理文件中规定“所购试药应符合现行药典有关要求”。 鉴于采购人员对药典的具体要求及变化可能不熟悉,申购部门应当在提出申购时注明试药的具体要求(如含量/纯度、水分等),并在验收时根据厂家提供的COA再次确认试药是否符合要求。
2.2 规范试药的贮存/使用/废弃管理
《8001试药》引入“化学品安全标签”和“化学品安全技术说明书”,为试药的包装与储存、使用及废弃处置提供科学指导。
企业可在“验收管理”环节中要求收集试药的化学品安全技术说明书(MSDS),以作为试药贮存/使用/废弃的参考文件。
2.3 规范试药的效期管理
《8001试药》提出“除另有规定外,试药及其制备的试液、试纸、缓冲液、指示剂与指示液应当关注有效性,必要或可行时,可通过制定有效期或采用灵敏度试验等方式予以保证”。
对于有效期的制定,中国药典并未给出具体细节。GMP指南仅提出“对于采购的试剂,应遵循生产企业规定的有效期;对于生产企业未规定有效期的试剂,使用单位可基于科学合理规定试剂有效期。”企业可参考GEON发布的《Recommendations on setting the expiry period for commercial and in-house-prepared reagents used in the laboratories of the OMCL Network》,制定公司内部的SOP,详细规定试药的有效期制定原则和策略。
同时,企业可通过“标签管理”的形式注明效期(包括未开瓶效期及开瓶后效期),并安排专人定期对过期试药进行废弃处置,以保证试药的有效性。
参考文献:
【1】2025年版中国药典二部凡例
【2】Chp2025《0861残留溶剂》
【3】Chp2025《0862 元素杂质》
【4】Chp2025《8001试药》
【5】《Recommendations on setting the expiry period for commercial and in-house-prepared reagents used in the laboratories of the OMCL Network》