前言
创新药,也称为原研药,是一个相对于仿制药的概念,指的是从机理开始源头研发,具有自主知识产权,具备完整充分的安全性有效性数据作为上市依据,首次获准上市的药物。按照药物研发的常规流程,一款药物从确定靶点到最后审批上市的整个研发周期通常耗时十数年的时间。专利保护期过后,就会被大量仿制。在美国食品药品监督管理局(FDA)的评审标准中,新分子实体(New Moleculer Entity,NME)和获得新生物制品许可的药物(Biologic License Application,BLA)均属于创新药。根据CFDA颁布的注册分类,分为五大类。本文中的新药特指1类新药。
新药研发模式
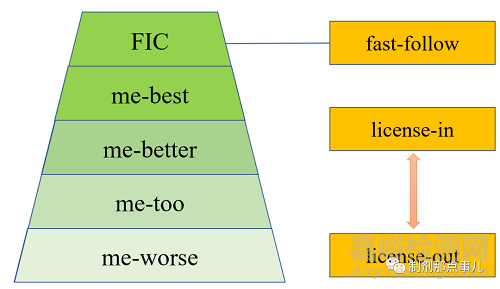
1类新药大致可分为以下几类(图1)。
▲ 图1-新药研发模式
“First in Class”即为首创新药。一般指能治疗某种疾病的新靶点、新机理药物;
“me-too”意为与FIC具有相似的作用机理和治疗效果的药物,;
“me-better”结构可能具有较大优化,在活性、代谢和/或毒性等各方面优于"me-too"药;
“me-best”即在同类创新药里最好;
“me-worse”则是相比“me-too”而言在疗效更差或毒副作用更强。
“fast-follow”指快速追踪新药的模式,以最快速度对“first in class”药物分子进行改造,也被称为“站在巨人的肩膀上依葫芦画瓢”;
“license-in”指购买授权许可;
“license-out”指出售授权许可。
药物发现阶段
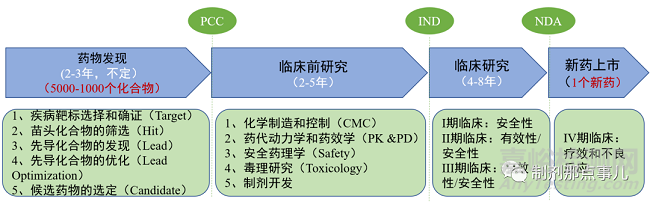
在生物医药研发领域,新药研发具有长周期、高成本、高风险的特点。业内人常说的“双十定律”即成功研发一款新药,即使过程比较顺利,也需要耗时十年、投资十亿美金,甚至更久更多。
1、疾病靶标选择与确证(Target)。
2、苗头化合物的筛选(Hit)。
3、先导化合物的发现(Lead)。
4、先导化合物的优化(Lead Optimization)。
▲ 图2-新药开发基本流程
IND申报及审批
在候选药物完成临床前研究后,便可向药品评审中心(Center for Drug Evaluation,CDE)(美国为FDA)提出临床试验申请(Investigational New Drug,IND)一般以IND作为新药研发的第二个关键节点。
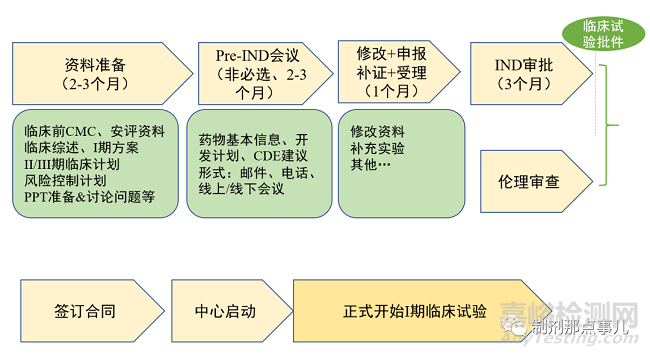
1、资料准备阶段。包括临床前研究的质量研究和安全性评价资料、临床试验方案、药效学/药代动力学/毒理学资料等(图3)。
▲ 图3-IND申报流程
2、Pre-IND会议。在正式向CDE提交IND之前,申请人可自愿选择是否进行Pre-IND会议(无偿),向监管机构提交产品的基本信息、开发计划,所需沟通交流问题的背景和数据,并提出拟解决问题的建议和想法等。对于缺乏新药注册的企业来说,这是保证成功的必要一步。
3、IND审批。CDE会在60天内通知申请人该新药物种是否可进行临床试验,若60天内未给出意见,则默认可以开始临床试验。
4、伦理审查。
5、签订合同。
6、中心启动会。临床试验正式开始前,必须在研究科室召开临床试验启动会,其目的了让所有参与临床试验的人员熟悉试验方案及具体操作流程。参与人员主要包括:研究者、申办方、机构人员、CRA和CRC等。经过启动会的当天准备、召开,待启动会结束后,即可正式开展I期临床试验。
药物临床试验
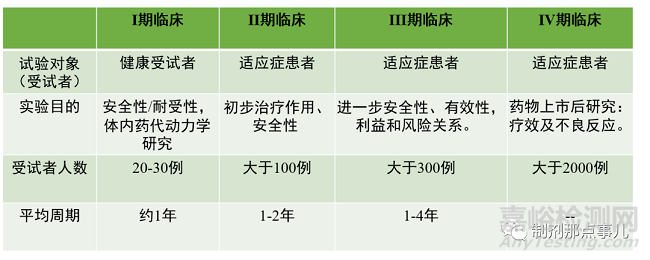
药物临床试验共分为四期,其中,I-III期临床试验为上市前,且每一阶段临床试验都会预设临床终点,达到临床终点方可开展下一阶段的试验;IV期临床试验为上市后研究。
▲ 图4-药物临床试验
1、I期临床试验。受试者为健康人。主要目的是:1)对药物的安全性及在人体的耐受性进行研究,考察药物副作用与药物剂量递增之间的关系;2)考察药物的体内药代动力学性质,包括代谢产物及代谢途径等。按照试验阶段可分为Ia期和Ib期。Ia期也称为单次剂量递增(SAD,Single ascending dose)试验,也是我们常说的“爬坡试验”。分析关键数据指标结果,直到达到预期水平。为制定接下来II、III期临床试验设计和给药方案提供依据。若药物增加新适应症,一般不需要再做I期临床试验。
2、II期临床试验。受试者为适应症患者。重点在于初步评价药物的安全性和疗效。应用安慰剂或已上市药物作为对照药物对新药的疗效进行评价,在此过程中对疾病的发生发展过程对药物疗效的影响进行研究;确定III期临床试验的给药剂量和方案;获得更多的药物安全性方面的资料,按阶段可分为IIa期和IIb期。IIa期(Proof of Concept,POC)即疗效探索,目的是证明药物的临床疗效和生物活性。这个阶段通常用于少数患者,主要疗效终点在给药后早期,评估其有效性;IIb期(Dose Finding,DF)即剂量确定,目的是确定显示生物活性,而副作用最小的最佳剂量,在这个阶段评估药物的疗效以及安全性,为了找到III期试验中最佳剂量,主要疗效终点在给药后后期。
3、III期临床试验。受试者为适应症患者,也被称为治疗作用确证(confirmatory)。目的进一步更大范围验证药物安全性和疗效,对药物的益处/风险进行评估。III期试验是最昂贵,最耗时和最困难的,试验设计通常是随机、对照、多中心的。可分为IIIa期和IIIb期。IIIa期试验是在药物的有效性被证明后,但在向监管机构提交注册申请之前进行的。研究的结果用于提交新药注册申请;IIIb期在提交注册申请后,但在获得药品批准并投入生产之前进行,目的是为了获得额外的安全性数据、发表文章、营销声明或准备药物上市。这也被称为上市前阶段(pre-marketing phase)。
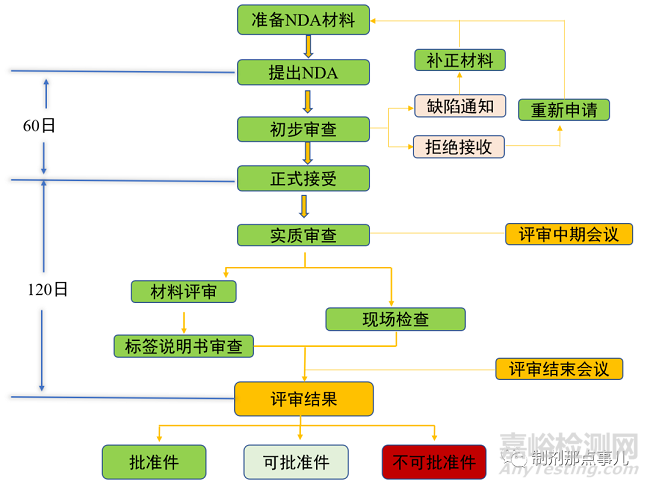
完成III期临床试验后,便可向国家药品监督管理局(National Medical Products Administration,NMPA)递交新药上市申请(New Drug Application,NDA)受理批准后便可进行新药上市(图6),一般以NDA作为新药研发的第三个关键节点。
▲ 图5-新药NDA流程
新药上市后
1、IV期临床试验。也被称为上市后研究(PMS,Post-Marketing Study)或上市后监测(PMS,Post Marketing Surveillance),主要目的是确定长期的安全性和有效性。可以在更长的时间和更大患者群体中对药物安全性进行进一步的监测和评估。
2、上市后再审批。一般上市后4-10年,主要是重新审核NDA中药物的安全性和有效性。