您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-05-30 12:18
浅谈生物类似药
以利妥昔单抗、雷珠单抗、非格司亭为例
由于药品研发具有周期长、投资大、风险高,因此,药物研发企业需要获得尽可能长时间的市场独占权,从而获得市场补偿,而专利制度就是市场独占的一个重要制度。医药行业对其专利保护的依赖性更强,专利在该行业中会影响64%的研发支出。各个国家都在建立符合本国国情的专利保护体系,以保护药品药品研发者的权益,提升行业药品创新的积极性。
但是,专利都存在有效期,一旦超过专利保护期,其他制药企业就可以生产这些药物,此时就出现了“仿制药”。由于没有像原研专利药一样高额的研发成本,同时不需要再做市场推广,仿制药的价格自然会比原研专利药低很多。一般来讲,仿制药的价格可以下降80%甚至更多。
01生物类似药的定义
由于生物药的特性以及不同国家和地区的监管要求和评审路径不完全相同,生物药的仿制药并不同与化学药的仿制药,且命名和定义也不尽相同。
1、根据中国药品监督部门《生物类似药研发与评价技术指导原则(试行)》,生物类似药是指:在质量、安全性和有效性方面与已获准注册的参照药具有相似性的治疗用生物制品。生物类似药候选药物的氨基酸序列原则上应与参照药相同。对研发过程中采用不同于参照药所用的宿主细胞、表达体系等的,需进行充分研究。
2、FDA:生物类似药(Biosimilar)是指和参照产品相比,尽管在非活性成分有差别,但是高度相似;在安全性、纯度和效价方面和参比药物没有显著差异。
3、EMA:生物类似药是包括EEA已批准的原研生物药活性成分变体的生物药物。需要基于全面的可比性研究,在质量特性、生物活性、安全性和效力方面建立与参比药物药品的相似性。
4、EHO:与已批准的参比生物治疗产品在质量上、安全性和效力方面均相似的生物治疗产品。
02生物类似药对医疗健康行业的意义
生物类似药对整个医疗健康行业的相关方,包括患者、医疗部门、医保部门、产业界等,都具有非常积极的意义,有利于提高各方的运营效率,实现社会整体福利的提高。
1、患者端:生物类似药的到来,将极大地提升众多高价特效生物药的临床可及性。更低的价格、更大的产量,意味着更多的病人可以支付得起。以中国为例,利妥昔单抗、阿达木单抗、曲妥珠单抗等生物药在临床的使用率远远低于西方发达国家,核心问题之一就是原研药高昂的使用成本限制了更多的病人,他们被迫只能接受疗效较差的传统治疗方案。生物类似药的出现,无疑将让这部分病人获益。以罗氏的贝伐珠单抗为例,其于2010年在中国获批,商品名为安维汀(Avatin)。2017年7月,罗氏安维汀通过谈判进入国家医保目录,从5223元/瓶降为1998元/瓶; 2019年,经医保谈判,再次降价24.92%,以1500元/瓶的价格被纳人国家医保。2019年12月,齐鲁制药的贝伐珠单抗注射液作为国内首个获批的Avasin生物类似药顺利上市,定价1266元/瓶,后降价为119元/瓶。2020年7月,信达生物的贝伐珠单抗注射液在某省的挂网价格为188元/瓶(4ml: 100mg)。
2、缓解医保支付压力:随着社会的发展和老龄化的不断加剧,我国医疗卫生支出上涨的压力持续增大。生物类似药的出现能够有效地引入竞争,大幅降低医保的支付压力,保持医保基金的运行安全和可持续性。
3、产业升级:由于生物类似药对原研药市场份额的不断侵蚀以及生物类似药自身价格在竞争中不断下降,产业界必须持续研发更多的新型药物,以保证其收入和利润的稳定。这相当于倒逼产业转型升级,加速产业的创新。
下表是生物药原研产品的专利到期时间和销售收入受生物类似药影响的情况。
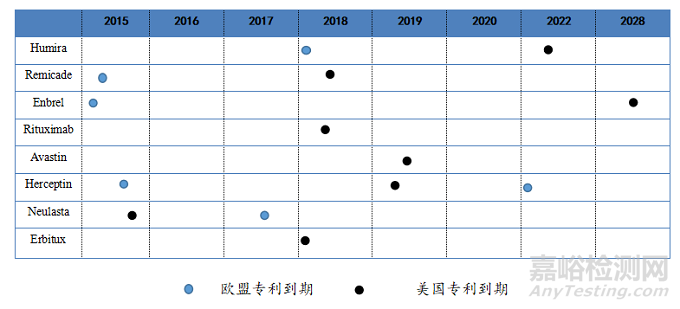
表1:部分生物药原研产品在欧盟和美国的专利到期时间
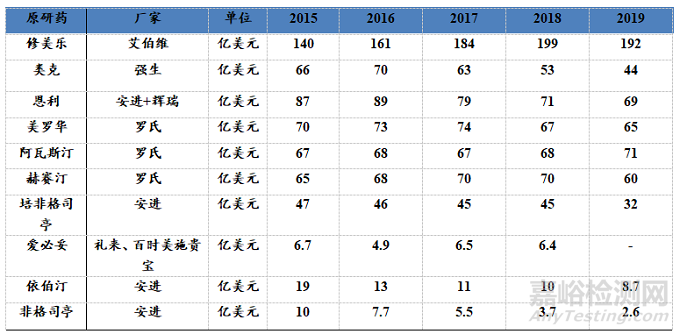
表2:生物原研药品种销售收入变化情况
03生物类似药的的研发流程与市场

图1 生物类似药的研发流程
对生物类似药的研发,首先要评估其质量属性和特性,然后将其与原研药进行比较研究。一般可分为三步:第一步对比质量研究;第二步对比非临床研究;第三步对比临床研究。
在第一步中,主要是对生物类似药的分子结构、生物活性等物理、化学和生物特性进行评估。由于基本不可能获得原研药的生产工艺,生物类似药开发的第一步是建立自行生产的工艺。由于生物技术产品的质量可能因制造方法的不同而受到影响,对生物类似药与其原研药的质量属性可比性评估至关重要。应在评估结果的基础上进行生产工艺的调整,直至生物类似药的质量属性尽可能地符合参照药的范围,并最终确保生物类似药和参照药在安全性和有效性方面没有临床意义上的差异。
第二步:主要包括动物药代动力学(PK)、 药效动力学(PD)和毒理学的评估,以应对由于生物类似药和参照药的质量属性存在一定的差异,评估这些差异对生物类似药的安全性和有效性的影响。
第三步:为证实生物类似药和参照药之间具有可比的临床疗效,通过该步骤,克服许多动物模型通常缺乏的必要灵敏度,评估人类的免疫原性。
总之,生物类似药的开发流程是一个逐步的、循序渐进的过程,是调整、适应、比较和确认的循环过程。只有在安全性、有效性和质量可控性方面与参照药之间没有临床意义上的差异时,生物类似药才可以被批准。
04中国生物类似药的市场情况
1、市场概况
中国的生物类似药起步较晚但在研数量最多,由于生物药的研发和生产壁垒较高,2019年之前中国尚未有国产生物类似药获批上市。2015年2月,中国药品监管部门制定发布了《生物类似药研发与评价技术指导原则(试行)》2019年2月25日,NMPA批准了复宏汉霖的利妥昔单抗注射液(商品名:汉利康)的上市,是首个中国“国产”生物类似药。随后,国产生物类似药陆续获批,2019年共获批4款,截至2020年9月,国内生物类似药共有8个获批上市,中国生物类似药迎来了突破性的进展。
从市场规模来看,中国生物类似药市场规模在不断扩大,2019年更是有了大幅度的增长。2019年全年销售额达到7900万元,2020年上半年的销售额更是达到9580万元。同时,2019年我国生物类似药的市场规模接近30亿元,增长幅度较大。随着中国生物类似药的监管审批和医保支付等路径越发清晰,生物类似药市场快速增长且潜力巨大。
2、价格形成机制
在中国未有生物类似药上市以前,生物药因其开发难度及专利等问题,市场直被跨国原研公司垄断。原研药可以单独定价,降价意愿不强,价格保持稳定。直到最近几年,原研药经过医保谈判后,价格大幅降低,才有资格进入国家医保目录。2017 ~ 2019年的三次医保准入谈判先后将31种创新生物制品纳人国家医保目录,谈判确定支付标准,并按照支付标准挂网采购。医保准入谈判已成为中国创新生物制品价格形成的主要控制措施,而生物类似药的上市进一步推动了这类药品的价格降低。具有生物类似药获批品种的生物制品也将不再纳人医保续约谈判范围,药品采购将替代医保谈判,成为控制其价格的主要举措。
05生物类似药药学研究的主要技术要求和评价基本原则
作为在质量、安全性和有效性方面与参照药具有相似性的治疗用生物制品,生物类似药的研发不仅需遵循生物药研发的一般流程,还需兼顾与参照药的相似性需求。根据中国药品监管部门于2015年发布的《生物类似药研发与评价技术指导原则(试行)》,生物类似药研发需要以比对试验研究证明其与参照药的相似性为基础,支持其安全、有效和质量可控。每一阶段的比对试验研究都应与参照药同时进行,并设立相似性的评价方法和标准。药学比对试验研究中应选择足够的代表性批次进行,并尽可能使用与参照药一致的、灵敏的、先进的分析技术和方法检测候选生物类似药与原研药之间可能存在的差异。生物类似药药学研究的主要技术要求和评价基本原则包括如下:
1、氨基酸序列需与参照药一致
生物类似药候选药物的氨基酸序列原则上应与参照药相同。在生物类似药研发早期,除了调研专利文献外,还应采用互补的技术手段对参照药的氨基酸序列进行表征和确认,如完整蛋白水平、亚基水平的分子量分析和肽图水平的氨基酸序列鉴定。另一方面,在无法获得参照药氨基酸序列信息的情况下,可以考虑开展测序分析实验,如使用基于质谱的从头测序方法或Edman降解法解析参照药的氨基酸序列。
2、生物类似药的理化性质和生物活性需与参照药相似,任何观察到的差异都必须充分证明不会影响药物的临床疗效和安全性
因为生物药固有的微观不均一性,不同批次生产的参照药以及生物类似药和参照药之间都不可能做到完全一致,或多或少会存在某些质量属性的波动或差异。根据可比性、相似性指导原则的要求,需要证明这些质量属性的波动或差异不会影响药物的临床疗效和安全性。
3、生物类似药产品中允许使用与参照药不同的辅料,但需证明包含的杂质和辅料都不会引起药效和安全性问题
中国药品监管部门的生物类似药评价指导原则要求进行处方筛选研究,并应尽可能与参照药致,对不一致的情况应有充足的理由。作为专利布局的一部分,参照药厂家通常会为其制剂申请专利。为了规避专利的影响,生物类似药厂家通常会使用不同的处方组成。和化学药不同,生物药对环境非常敏感,辅料和初级包装的变化均有可能影响产品的稳定性,在选用与参照药不同的制剂处方时须详细评估相关影响。
4、中国和国际药监机构的生物类似药药学研究指南遵循相同的原则和相似的技术要求,但在操作细则上存在差异
中国药品监管部门、WHO、EMA和FDA监管下的生物类似药学研发都遵循“头对头”的比对研究原则和逐步通进的研发次序,有着相似的技术要求。但在操作细节上,如参照药批次的选择、多照药的储存条件、分析方法的选择标准以及相似性评价方法上存在差异(表1)。
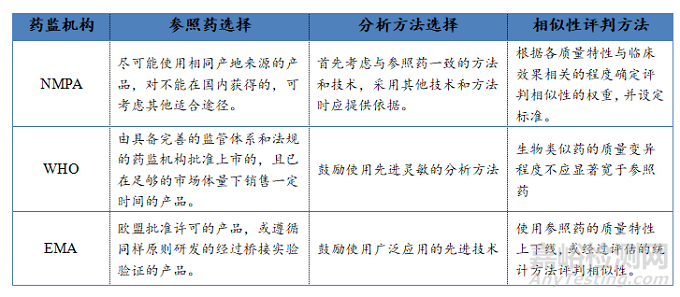
表3:各种审评机构对生物类似药药学研究要求细则比较
06生物类似药的审评审批政策
1、国际上生物类似药的相关政策
(1)美国
在美国,生物类似药的临床试验申请与创新生物制品的申报流程相同,同样遵循临床研究申请(IND)。
上市申请的主要流程为:
<1>申请前会议:FDA强烈建议申请人在BPD-351(k)BLA会议上与相应的审评部门讨论计划申请的内容。此会议将由FDA审评小组出席,包括相应专业的FDA高级职员。
<2>初次申请提交:FDA希望申请人根据BPD-351(k)BLA会议上双方达成的一致内容进行初次申请的提交。
<3>发出第74天信函:FDA将遵循在“第74天信函”中规定的程序,即对申请人在初次申请审查中发现的实质性审查问题进行确认和沟通。如果在申请审查期间未发现实质性审查问题,FDA将通知申请人。
<4>审评时限:FDA的目标是实现对90%的申请在受理后的10个月内完成。
<5>中期沟通:FDA于2018年6月发布了生物类似药正式会议指南《行业指南:FDA与生物类似药申办方之间的正式会议》,在审评过程中根据讨论问题的不同,申请人可向FDA提出会议沟通申请。
<6>晚期会议和咨询委员会会议:FDA一般在目标批准日期之前2个月内开展咨询委员会会议。晚期会议是在咨询委员会会议之前不少于12个日历日开展。
<7>检查:递交申请10个月之内完成GCP、GLP以及GMP检查。
另外,按照生物类似药付费法案(BsUFA II)的要求,还会由独立的第三方对生物类似药审评过程中的效率进行评估,主要目标是审查351(k)申请的审评效率和效果,减少审批所需的审评周期。
(2)日本-按照新药程序注册申请
<1>临床试验申请
日本对药物临床试验的审评机制采取30 天默示许可方式。当申请人进行首次临床试验申请时,为了防止受试者健康危害的发生,PMDA可就该申请开展必要的审评(首次申请为“30 天审查”,非首次为“14 天审查”)。临床试验申请由厚劳省受理,技术审评由PMDA负责。
在获得厚劳省默认许可后提交伦理委员会审查,不可同步进行;决策形式为默认许可,不发批准函;临床试验被暂停后,由申办方提交修正后的临床试验方案,经与PMDA沟通并通过伦理委员会审查后,方可再次启动。
<2>上市申请
所有医疗用药品的上市许可均按以下程序审评审批:
a)申报前可与PMDA进行面对面沟通交流;
b)申请进行GLP检查及GLP认证,获得GLP评价结果通知书及GLP认证书;
c)向PMDA提交申报资料;
d)申请进行GCP实地和文档是否符合相关标准的核查;获得GCP实地检查结果通知书及文档资料检查符合标准通知书
GCP检查目的是检查该药的临床试验是否符合GCP标准,分为现场检查和资料审查,由PMDA下属的“合规和标准办公室”负责。现场检查是对试验的原始数据(如原始病历、实验室检查报告、患者日记卡等)和病例报告表(CRF)之间的一致性进行检查,检查对象分为研究者和申办者。资料审查不仅是对临床部分的检查,也包括对药品质量(主要是药品检验和稳定性试验)和非临床试验部分的检查。其中,临床部分是对CRF和注册资料之间的一致性进行检查,检查对象是申办者。
e)技术审评
技术审评的程序包括:PMDA组织申请人与审评员和核查员的沟通交流;以PMDA为主体进行的合规性核查;由审评团队起草审评报告;召开审评员与外部专家的审评专题会;如需要,由审评部长主持召开与申请人的沟通交流会;根据需要,审评员与外部专家再次召开审评专题会;完成审评报告;形成审评结论,制作审评结论通知书。
f)行政审批
审评结果和审评通知书报送厚生省;厚生省咨询药事食品卫生审议会;厚生省发布批准/不批准通知书。
(3)韩国-临床试验及上市注册申请程序
<1>临床试验申请
申报临床研究申请时,申请人将CMC数据、稳定性数据、药理学/毒性数据、临床方案等提交给临床试验管理部门;由生物制品部进行审评,审评完成后,要求补充额外信息的,补充资料需在一个月内提供;审评通过的,将由临床试验管理部门通知申请人研究开始,至研究完成后接受检查;申请人提供的数据不足以支持研究的,临床试验须暂停,整个审评过程共计30 天。
<2>上市申请
生物类似药的上市许可申报和审评按照基于数据评估的药品的流程进行。申请人提交上市申请至生物制品部,包含CMC数据、稳定性数据、非临床数据、临床数据以及登记状态等信息;生物制品部对资料进行审评,如有需要,将进行咨询委员会会议;审评完成后,如要求补充额外信息或修改要求的,资料需在2个月内提供;审评通过的,进行标签审查及GMP检查,通过后,通知申请人上市申请已通过,整个审评过程共计115 天。
(4)欧盟
生物类似药临床试验许可和监督由各成员国负责。将欧盟临床申请制度与美国进行比较,两个监管机构存在一些差异,具体如下:<1>欧盟对临床试验进行全面评估,对每个临床试验方案,都需要全面评估和批准一个新的临床试验许可(CTA),此过程必须提供所有的质量、安全性和有效性数据。美国对于相同的治疗领域的其他临床方案可以通过简单的流程在同一个IND申请中提交,而无需重新提交所有的初始IND申请信息。<2>欧盟人用药品委员会(CHMP)给予申请人的科学建议与临床试验申请的国家审评机构之间未建立有效的联系。如申请人可以遵循CHMP的建议进行研究设计,然而评估临床试验方案的国家审评机构可能不了解此建议,并且有可能会要求申请人采取与CHMP的建议有冲突的方法。相反,在美国,一旦启动IND申请,所有临床试验活动和研发讨论将在IND中提交,申请人和监管机构都可以充分了解产品的研发历史以及如何根据建议设计临床试验。
大多数生物类似药在内的以生物技术生产的生物药品上市申请,由EMA负责评估。此类产品是通过涵盖所有欧盟成员国和欧洲经济区国家的集中程序进行评估的,之后出具单一批件。由所分配的国家管理局进行评估,即主审评员(Rapporteur)和副审评员(Co-rapporteur)进行,他们代表整个欧盟进行科学评估。审评员提出问题由CHMP审评,之后可以添加问题并对问题重要性升级或降级。最终CHMP对审评员提出的科学建议进行投票并决定是否给出“肯定”的推荐批准或“否定”的拒绝建议。根据CHMP的技术审评建议,EC负责最终发布适用于所有欧盟成员国且有约束力的上市许可。欧盟委员会在科学事务上须遵从CHMP,因此EC通常遵循CHMP意见。
<1>集中程序
集中程序遵循明确的时间表。根据时间表申请人可以预估什么时间监管机构提出问题。审评根据适用的技术指南对提交的数据进行评估。对于生物类似药,重点是通过与参照药“头对头”的比对结果审评整个证据链,从而考察是否建立生物相似性。如果该申请人在研发产品期间进行了科学咨询,管理局将审评申请人对科学建议的遵循程度以及未遵循科学建议的理由是否合理。
审评还评估GCP和GMP的合规性,根据需要开展检查。当程序结束时如仍然存在问题,并且无法以书面形式解决疑问时,该程序还可选择“口头解释”。在这些情况下,申请人将与CHMP举行正式会议。如被要求进行口头解释,则在流程的第181天进行,并于第210天或之前发布最终CHMP意见。需要说明的是,口头解释在形式上与美国同类咨询委员会有很大的不同,申请人在本次会议上的发言仅有30 min左右。
EMA将不会对生物制品样品检验作为上市许可审评的一部分。生物制品需要申请人将成品在欧洲经济区内由有资质的机构人员进行检测和批量发放。EMA需要申请人提交包装模型和最终包装样本,但不需要提交样品。对于所有经批准的产品,EMA还会制定抽样和检验计划,以验证市场上经集中审评许可的药品的质量,并检查其是否符合批准的质量标准。
欧盟委员会在发布产品批准的决定后,EMA制定欧洲公共评估报告(EPAR),经申请人确认后,EPAR将在EMA网站上与产品特征概要(Summary of product characteristics,SmPC)以及标签包装说明书一起发布。EPAR包含EMA审评摘要以及他们是如何达成决定的。
<2>复议
如果申请人不同意CHMP的否定性意见,申请人可以提出上诉,这称为“复议”。复议的流程大致如下:15天请求复议;60 天提交理由;CHMP 60 天重新评估最初的审评意见,过程中不允许提交新的数据;可以咨询科学建议小组。
2、中国关于生物类似药的有关政策
(1)《生物类似药研发与评价技术指导原则(试行)》2015年
本指导原则所述生物类似药是指:在质量、安全性和有效性方面与已获准注册的参照药具有相似性的治疗用生物制品。生物类似药候选药物的氨基酸序列原则上应与参照药相同。对研发过程中采用不同于参照药所用的宿主细胞、表达体系等的,需进行充分研究。本指导原则适用于结构和功能明确的治疗用重组蛋白质制品。对聚乙二醇等修饰的产品及抗体偶联药物类产品等,按生物类似药研发时应慎重考虑。
研发和评价的基本原则有:
l 比对原则
生物类似药研发是以比对试验研究证明其与参照药的相似性为基础,支持其安全、有效和质量可控。每一阶段的每一个比对试验研究,均应与参照药同时进行,并设立相似性的评价方法和标准。
l 逐步递进原则
研发可采用逐步递进的顺序,分阶段证明候选药与参照药的相似性。根据比对试验研究结果设计后续比对试验研究的内容。对前一阶段比对试验研究结果存在不确定因素的,在后续研究阶段还必须选择敏感的技术和方法设计有针对性的比对试验进行研究,并评价对产品的影响。
l 一致性原则
比对试验研究所使用的样品应为相同产地来源的产品。对候选药,应当为生产工艺确定后生产的产品,或者其活性成分。对工艺、规模或产地等发生改变的,应当评估对产品质量的影响,必要时还需重新进行比对试验研究。
比对试验研究应采用适宜的方法和技术,首先考虑与参照药一致,对采用其他敏感技术和方法的,应评估其适用性和可靠性。
l 相似性评价原则
比对试验研究所使用的样品应为相同产地来源的产品。对候选药,应当为生产工艺确定后生产的产品,或者其活性成分。对工艺、规模或产地等发生改变的,应当评估对产品质量的影响,必要时还需重新进行比对试验研究。
比对试验研究应采用适宜的方法和技术,首先考虑与参照药一致,对采用其他敏感技术和方法的,应评估其适用性和可靠性。
对临床比对试验研究结果判定为相似的,可按本指导原则进行评价。
(2)《接受药品境外临床试验数据的技术指导原则》2018年
随着药物研发全球化进程的加速,越来越多的跨国公司和国内企业通过开展国际多中心临床试验用于支持全球的注册申请。药品境内外同步研发可减少不必要的重复研究,加快药品在中国上市的进程,更好满足患者的用药需求。为进一步落实《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号),发布《接受药品境外临床试验数据的技术指导原则》(以下简称《指导原则》)。
本技术指导原则旨在为境外临床试验数据用于在我国进行药品注册申请提供可参考的技术规范,以鼓励药品的境内外同步研发,加快临床急需、疗效确切、安全性风险可控的药品在我国的上市,更好满足我国患者的用药需求。本指导原则适用于已有境外临床试验数据的药品在中华人民共和国境内的各类注册申请。
提交在境外获得的临床试验数据的主体,应当符合《药品注册管理办法》对于注册申请人资质的要求。例如,国内企业在境外开展的临床研究所获得的数据,符合我国相关法规、技术规范要求的,可以用于在我国的注册申请。
鼓励开展境内外同步研发。境外临床试验数据可以用于支持需要进行有效性和/或安全性评价的各类注册申请。例如,在境外开展的早期临床研究数据,可用于支持在我国开展注册临床试验的申请。
境外开展的临床试验数据用于支持在我国药品注册的,其研究质量应当不低于本技术指导原则的标准。例如,在药物临床试验管理规范与ICH标准相差较大的国家或地区完成的临床试验,或早年完成的临床研究,应当有充分的依据证明其研究质量符合相关要求,必要时接受国家药品监督管理局的核查。临床试验数据的整理要求增加按照“《药品注册管理办法》的要求”
l 境外临床试验数据的提交形式
用于在中国进行注册申报的境外临床试验数据,应提供中文翻译件及其原文。采用CTD格式提交临床试验数据,应当符合《关于适用人用药品技术协调会二级指导原则的公告》(2018年第10号)的要求。
l 境外临床试验数据的评价
境外完成的药品临床试验数据用于在我国进行药品注册申请,首先应当确保研究质量,研究数据应真实、完整、准确和可溯源。
境外临床试验数据的可接受性评价基于药品本身的作用特点和相关技术指导原则开展。考虑的因素包括与遗传和生理以及文化和环境特性相关的因素,如流行病学、病因、药物作用机理、药物代谢酶的基因多态性、医疗环境、文化差异等因素。例如,境外临床试验中的海外中国人数据,当药物主要经基因多态性酶代谢,且药物代谢对疗效和/或安全性有关键性影响,可以作为人种差异分析的主要因素;而对于代谢过程受饮食影响较大的药物,或所治疗的疾病受环境因素、文化、医疗实践影响较大的,则不能作为人种差异分析的主要因素。
境外完成的仿制药生物等效性试验数据,研究质量好,具备真实性、完整性、准确性和可溯源性的,也可用于在我国的注册申请。
生物类似药的境外临床试验数据质量也应当满足真实、完整、准确和可溯源的要求,其技术要求与创新药和仿制药均不完全一致,应结合具体情况具体分析。
(3)《药品注册管理办法》2020年修订
生物制品注册按照生物制品创新药、生物制品改良型新药、已上市生物制品(含生物类似药)等进行分类。境外生产药品的注册申请,按照药品的细化分类和相应的申报资料要求执行。国家药品监督管理局药品审评中心(以下简称药品审评中心)负责药物临床试验申请、药品上市许可申请、补充申请和境外生产药品再注册申请等的审评。
使用境外研究资料和数据支持药品注册的,其来源、研究机构或者实验室条件、质量体系要求及其他管理条件等应当符合国际人用药品注册技术要求协调会通行原则,并符合我国药品注册管理的相关要求。
对于创新药、改良型新药以及生物制品等,应当进行药品注册生产现场。
图1:药品注册生产现场核查和上市前药品生产质量管理规范检查
(4)《M4:人用药物注册申请通用技术文档(CTD)》2016年
本指导原则主要适用于新药(包括生物制品)的注册申请过程中需要提交资料的组织架构信息。本指导原则并非说明需要开展哪些研究,而仅说明对所获得的数据进行呈现的适当格式。
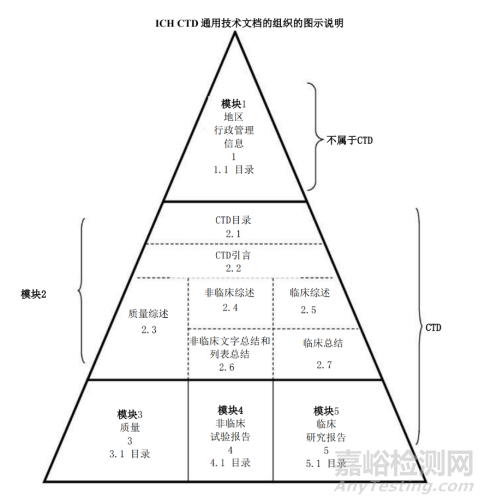
图3:ICH CTD通用技术文档的组织的图示说明
07案例:中国生物类似药研发管线中的利妥昔单抗、雷珠单抗和非格司亭
1、利妥昔单抗
(1)产品概况:利妥昔单抗注射液(rituximabinjection)是由基因泰克和BiogenIdec公司合作开发的一种采用基因工程技术合成的人鼠嵌合IgG1 K单克隆抗体,靶向B细胞表面的CD20跨膜蛋白。利妥昔单抗联合化疗是CD20阳性非霍奇金淋巴瘤的治疗金标准,大量随机临床试验及长期随访数据表明,在标准淋巴瘤化疗方案中加人利妥昔单抗,可以显著提高CD20阳性滤泡性非霍奇金淋巴瘤和弥漫性大B细胞淋巴瘤患者的生存率以及治疗效率。
(2)作用机制:CD20是一种在免疫系统中的前体B细胞及成熟B细胞的表面都广泛表达的蛋白质,其恶变与B细胞淋巴瘤、白血病、免疫疾病和炎症疾病等相关。利妥昔单抗可与B细胞表面的CD20相结合,通过细胞调亡、直接的生长抑制作用、补体依赖的细胞毒性( CDC)及抗体依赖细胞介导的细胞毒性(ADCC) 启动免疫应答,最终清除B细胞,从而降低异常B细胞的作用。
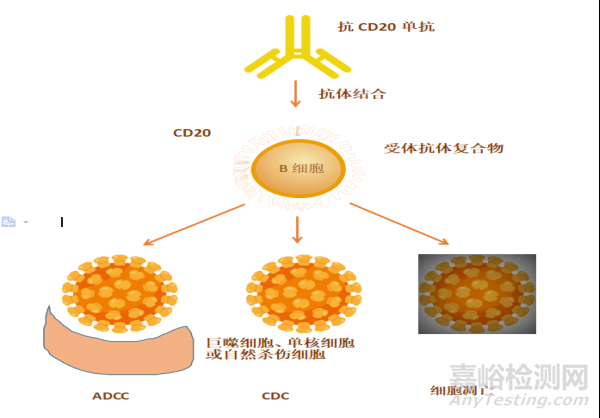
图4:利妥昔单抗作用机制示意图
(3)原研情况:利妥昔单抗 (Mab Thera) 于1997年获美国FDA批准而上市,1998 年在欧洲获得批准,2000年获批进入中国,在中国的商品名为美罗华。目前,利妥背单抗在中国获批可用于:①复发或耐药的滤泡性中央型淋巴瘤;②先前未经治疗的CD20阳性I~IV期滤泡性非霍奇金淋巴瘤;③CD20阳性弥漫性大B细胞非霍奇金淋巴瘤( DLBCL);④慢性淋巴细胞白血病(chronie lymphoeytic leukemia.CLL);⑤初治滤泡性淋巴瘤(ollicular lymphoma, FL)患者经利妥昔单抗联合化疗后达完全或部分缓解后的单药维持治疗。根据IQVIA CHAPTM的数据统计,2018 年和2019年利妥昔单抗在中国境内的销售金额分别约为20. 7亿元人民币和25.03亿元人民币。
(4)专利情况:利妥昔单抗注射液原研产品在美国、欧洲和中国的专利已分别于2015年、2013 年和2013年到期。
(5)生物类似药开发情况:2019年2月22日,国家药品监督管理局批准上海复宏汉霖生物制药有限公司研制的利妥昔单抗注射液(汉利康)的上市注册申请。作为国内首个按照《生物类似药研发与评价技术指导原则(试行)》研发和申报生产的生物类似药,汉利康一改我国生物药市场历史格局,开启了中国生物类似药时代。截至2020年2月,仅复宏汉霖的汉利康和信达生物的利妥昔单抗生物类似药获批上市:信达生物的IBI -301已提交新药上市已获批;神州细胞的SCT400已提交上市申请,另有海正药业、鼎康生物、正大天晴等公司的该药物处于三期临床阶段。全球范围内,截至2020年9月,Celtrion/ Teva的Truxima、诺华的Riximyo和辉瑞的Ruxience 共3款利妥昔单抗生物类似药已获得美国FDA和欧盟EMA的批准。
(6)适应症开发情况:截至2020年9月,原研利妥昔单抗在全球范围内获批的适应症包括非霍奇金淋巴瘤、白血病等血液肿瘤疾病以及类风湿关节炎、寻常型天疱疮等自身免疫性疾病。在国内,利妥昔单抗针对类风湿关节炎的适应症暂未获批,但与已上市的其他抗类风湿关节炎制剂相比,利妥昔单抗具有给药频次低、药物有效性持续时间长等优势,可大幅提升患者用药依从性,从而改善患者的生活质量、降低患者的医疗负担。目前,复宏汉霖针对类风湿关节炎这一适应症的开发已进人I期临床阶段,有望为国内类风湿关节炎患者提供更加优质、高效的治疗选择。利妥昔单抗在美国、欧洲和中国已批准和正在开发的适应症如表4所示。
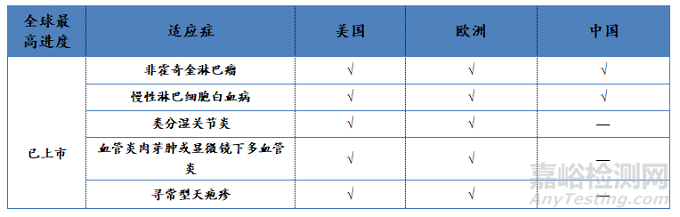
表4:利妥昔单抗在开发情况
2、雷珠单抗
(1)产品概况:雷珠单抗由罗氏子公司基因泰克和诺华联合开发,是一种人源化IgGlk型单克隆抗体的Fab片段,能与血管内皮生长因子A (VEGF-A)活化形式的受体结合位点相结合,阻止VEGF-A与内皮细胞表面的VEGF受体( VEGFR-1和VEGFR-2)结合,从而减少肿瘤的新生血管生成。该药用于治疗新生血管(湿性)年龄相关性黄斑变性( AMD)、视网膜血管阻塞(RVO)引起的黄斑水肿、糖尿病性黄斑水肿( DME)和糖尿病性视网膜病变(DR)。
目前,国内有2家的该药物处于临床三期,1家处于临床一期,2家处于临床前研究阶段。其中,齐鲁制药的QL-1205于2016年9月获得临床批件,目前已经到了临床三期阶段,是研发进度最快的雷珠单抗生物类似药。
(2)作用机制:雷珠单抗与贝伐珠单抗的作用机制相同,能够特异性结合VEGF- A。VEGF- A能使血管生长并渗漏组织液和血液,产生黄斑损伤。通过阻断VEGF-A,雷珠单抗可以减少眼底处血管的生长,并控制血管的渗漏和肿胀。雷珠单抗在结构上去掉了Fc片段而仅保留抗体的Fab片段,这一改变具有以下优势:①能够特异性结合VEGF-A;②分子量更小,具有更好的组织穿透性;③消除ADCC和CDC对安全性的影响。原研情况雷珠单抗商品名为Lucentis@ ( 诺适得),下称Lucentis或诺适得,于2006年6月30日获得美国FDA批准。目前,该药物获批的适应症包括新生血管( 湿性)年龄相关性黄斑变性、视网膜静脉阻塞后黄斑水肿、糖尿病性黄斑水肿、糖尿病视网膜病变和近视性脉络膜新生血管。2007 年,该药物获得欧洲EMA批准上市,用于治疗新生血管(湿性)老年性黄斑变性、糖尿病性黄斑水肿所致视力损害、增殖性糖尿病视网膜病变、视网雅静脉阻塞(简称RVO,包括分支静脉阻塞( BRVO)和中央静脉阻塞(CRVO))] 继发黄斑水肿所致的视力损害和脉络膜新生血管所致的视力损害。2012 年4月,该药物在国内获批上市,被批准用于治疗湿性老年性黄斑退化症; 2018年5月,被批准用于治疗继发于视网膜静脉阻塞(包括BRVO和CRVO)的黄斑水肿引起的视力损害; 2018 年11月,被批准用于治疗脉络膜新生血管[ CNV,包括继发于病理性近视(PM)的和其他原因引起的导致的视力损害。至此,诺适得成为目前国内唯一拥有此三项适应症的药物。
Lucentis的全球销售额为: 2016-2020年分别为32.55亿美元、33.2亿美元、37.42亿美元、39.26亿美元、33.77亿美元。国内重点医院的采购金额(来自IMS数据库)2016-2019年分别为3.4亿元人民币、3.9亿元人民币、5.0亿元人民币、6.2亿元人民币。
(3)专利情况:序列专利保护已于2018年到期。
(4)生物类似药开发情况:目前国内有四家按照生物类似药在进行开发。齐鲁制药在开展国际多中心的三期临床研究;联合赛尔的该药物于2019年1月被批准临床,按2类申报,申报的药品名称为雷珠单抗;杰科生物和杭州中美华东两家按照15类申报临床。欧美在开发生物类似药的有三家,分别是Biog CmbH公司、三星生物和Stada公司,2019年均在开展三期临床研究。
适应症开发情况截至目前,全球获批的适应症包括脉络膜新血管生成、湿性老年性黄斑退化症、视网膜静脉阻塞、糖尿病性黄斑水肿、糖尿病性视网膜病变和早产儿视网膜病共6个。目前,雷珠单抗在美国、欧洲、中国和日本的批准和开发情况见表5。
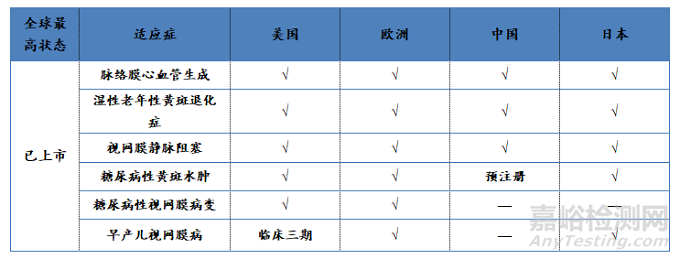
表5:雷珠单抗适应症开发情况
3、粒细胞集落刺激因子
(1)概况:粒细胞集落刺激因子(Granulocyte Colony Stimulating Factor,G-CSF)是刺激嗜中性粒细胞祖细胞和成熟中性粒细胞存活、增殖、分化和活化的糖蛋白,它在抗感染的非特异性细胞免疫过程中起重要作用。G-CSF临床主要用于预防和治疗肿瘤放疗或化疗后引起的白细胞减少症、治疗骨髓造血机能障碍及骨髓增生异常综合征、预防白细胞减少可能潜在的感染并发症等,目前市场上已经上市的G-CSF有非格司亭(Filgrastim)、培非格司亭(Pegfilgrastim)、利培非格司(Lipegfilgrastim)以及非格司亭生物仿制药。长效G-CSF相对于短效产品具有更好的临床获益,因此逐渐替代短效G-CSF的市场。
预计2019年国内长效G-CSF的市场空间约为101亿元,且随着肿瘤患者人数的增长以及用药渗透率的提高,这个数值仍在增长。假设年 增速为3%,则2027年国内长效G-CSF市场空间为127.9亿元,目前长效G-CSF的渗透率为36%,并且处在高速增长过程中,渗透率持续提高。预计到2027年渗透率有望提高到50%, 则届时市场规模为64亿元。根据在研进程,预计2026年内国内长效G-CSF的厂家大概在6-7家,平均每家市占率在14%-17%。

图5:粒细胞集落刺激因子迭代产品发展历史
(2)产品分类:<1>非格司亭(Filgrastim):是应用重组DNA技术生产的甲硫氨酸人粒细胞集落刺激因子,包含175个氨基酸残基,其氨基酸序列与人的DNA序列预测的天然序列相同,仅仅是在N端插入了甲硫氨酸。非格司亭由Amgen和日本协和发酵麒麟株式会社(Kyawa Hakko Kirin)共同研发。1991年2月20日获得美国FDA批准上市,商品名为Neupogen,1991年10月4日获得获得日本PMDA批准上市,商品名为Gran。FDA批准Teva公司的tbo-filgrastim(tbo-粒细胞集落刺激因子,又名XM02-filgrastim)于2012年8月29日上市,商品名为Granix,该药为监管部门批准的首个重组filgrastim生物仿制药。另一个被FDA批准上市的非格司亭生物仿制药为瑞士制药巨头诺华(Novartis)旗下Sandoz的Zarxio®(filgrastim-sndz),于2015年3月6日获批。该药是通过美国《生物制品价格竞争与创新法案》(BPCIA)创建的新的生物仿制药途径批准的首个生物仿制药,是美国有史以来第一个真正意义上的生物仿制药。
<2>培非格司亭(Pegfilgrastim):是Amgen公司开发的非格司亭的长效剂型,通过对粒细胞集落刺激因子(G-CSF)进行聚乙二醇(PEG)修饰,延长在体内的代谢时间,从而达到更好的疗效,临床上主要用于减少化疗相关的中性粒细胞减少症发生率,一个化疗周期中只需给药1次。聚乙二醇(PEG)是中性、无毒、无免疫原性、有良好的生物相容性的高分子聚合物,偶联到蛋白分子表面(PEG化),可改变其生物分配行为和溶解性,产生空间屏蔽,减少药物的酶解,避免在肾脏的代谢中被快速消除,并且可以降低或者消除诱导产生中和抗体和与抗体结合的能力,因而被广泛用于药物的修饰。培非格司亭于2001年1月31日首次获得美国FDA批准,2002年8月22日获得EMA批准,商品名Neulasta®和Ristempa®,2014年9月26日获得PMDA批准上市。Amgen在美国和欧洲市场销售,日本协和发酵麒麟株式会社在日本市场销售。非格司亭的仿制药市场竞争异常激烈,已经严重影响了非格司亭原研药企的销售额。近年来非格司亭的销售额下滑严重。而培非格司亭自2001年上市,销售额一路攀升,2003年的销售额就已达到13亿美元,远远超过了非格司亭,近年来其销售额更是突飞猛进,2015年其销售额已达到48亿美元。
培非格司亭的核心专利于1989年以国际专利申请,即PCT申请提交(公开号WO9006952A1),进入了大部分药品主流市场,并陆续在欧洲、美国、日本获得授权。美国专利US5824778A由于专利补偿制度,专利到期日为2015年10月20日。而欧洲和日本的专利权早在2009年就已经到期。值得注意的是培非格司亭的核心专利并没有进入中国。该专利独立权利要求保护了“至少一分子聚乙二醇(PEG)偶联的具有生物活性的G-CSF”。说明书中重点介绍了培非格司亭的合成方法,采用定点修饰,修饰的位点是蛋白中第1位半胱氨酸的α-氨基和赖氨酸侧链氨基,而PEG采用直链型。
<3>利培非格司亭(Lipegfilgrastim):是一种新颖的、聚乙二醇化、糖基化的长效型非格司亭。酶的合成技术具有区域选择性和立体选择性的优势,通过糖基转移酶的作用可以将PEG修饰的糖直接转移至多肽的主链上,从而实现在体外生产特异性修饰的,均一的生物治疗剂。利培非格司亭则是基于上述糖聚乙二醇化(GlycoPEGylation)技术产生的一种长效重组粒细胞集落刺激因子(G-CSF)。最初由Neose Technology公司(主要研究领域为糖基化)研发,随后Ratiopharm Group获得其开发权,2010年Teva将其收购并最终将利培非格司亭收入囊中,利培非格司亭于2013年7月25日获得EMA批准上市,商品名为Lonquex®。利培非格司亭将成为Amgen非格司亭(Fligrastim)和培非格司亭(Pegfilgrastim)强有力的竞争对手。
该专利于由Neose Technologies提出,于2004年12月3日以国际专利申请,即PCT申请提交(公开号WO2005055946A2),目前已经在日本、中国、欧洲获得授权,专利到期日为2024年。美国专利US20070254836A1处于公开状态。
<4>硫培非格司亭(Mecapegfilgrastim):为一种水溶性聚合物修饰的G-CSF偶联物,同传统的偶联物和方法相比,培非格司亭采用创新聚乙二醇(PEG)修饰蛋白技术。其适应症为:非骨髓性癌症患者在接受容易引起发热性中性粒细胞减少症发生的骨髓抑制性抗癌药物治疗时,降低以发热性中性粒细胞减少症为表现的感染发生率。从硫培非格司亭获批时递交的临床资料来看,在乳腺癌患者中其升白效果优于惠尔血(重组人粒细胞刺激因子注射液,安进)。在非小细胞肺癌患者中的升白效果远优于安慰剂组。
根据《柳叶刀》杂志统计,中国每年新增癌症患者数量457万例,需要预防中性粒细胞减少 (化疗之后白细胞减少)的患者总体占比超10%。长效GCSF升白针是预防和治疗中性粒细胞减少的标准疗法。从全球市场格局来看,长效G-CSF后来居上占据主导地位,市场份额超过70%,代表药物为安进公司的Neulasta(聚乙二醇非格司亭),该药2017年销售额为46.48亿美元。在中国,预计2021年GCSF升白针市场规模约75亿元RMB,其中长效GCSF升白针市场规模>50亿元RMB。之前由于医保等诸多原因,国内长效G-CSF占比不足20%。2017年医保目录调整,聚乙二醇重组人粒细胞刺激因进入国家医保乙类目录,国内长效GCSF进入快速发展期。至2021年9月,国内升白针市场已有的和可能的竞争对手包括:①石药集团(01093),培非格司亭生物类似药(津优力),纳入医保支付。②齐鲁制药,培非格司亭生物类似药(新瑞白),纳入医保支付。③恒瑞医药(SH600276),硫培非格司亭(19K),纳入医保支付。④普那布林,已经提交中美两地上市申请NDA
资料来源:
1.生物类似药:从研发到使用/沈阳药科大学亦弘商学院主编.—北京:中国医药科技出版社,2021.6
2.雪球https://xueqiu.com/
3.生物类似药研发与评价技术指导原则(试行)
4.接受药品境外临床试验数据的技术指导原则
5.《药品注册管理办法》
6.《M4:人用药物注册申请通用技术文档(CTD)》
7.腾讯网:法规角度对生物类似药审评审批的考量
8.中泰证券《生物类似药:仿制药中的“创新药” ——生物类似药专题报告》2017.6
9.国金证券《生物类似药:全球方兴未艾,中国奇点临近!》2018.6
10.平安证券《生物类似药市场蓬勃发展,单抗类似药执牛耳》2017.6
11.药渡:从非格司亭的迭代产品看生物药的发展方向
12.西南证券《从国内外Neulasta及其Biosimilar看F-627市场》2020.7

来源:会会药咖


