您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2021-07-22 20:09
在医学诊断、检查与治疗中,经常会用到医用手套,医用手套分为无粉和有粉两种,其中有粉手套可以方便医生穿脱,但也就是这种方便穿脱的有粉手套被FDA发布禁令自2017年1月18日起正式全面禁止,也就是说这类手套在美国可能无论如何无法获得FDA的批文进行上市销售。到底是什么原因导致的禁止?以及背后有什么故事,我们一起来说道说道。
(一)有粉手套什么时候被FDA禁止的?
美国医疗器械禁令是指全面禁止医疗器械当前和未来的销售、分销和制造。根据《联邦食品、药品和化妆品法》第516(a)节,及21 CFR 895.20,如果FDA根据所有可用的数据和信息发现该器械对患者或用户关于该器械的好处构成重大欺骗,或不合理且重大的疾病或伤害风险,且无法通过更改标签来纠正,FDA有权禁止用于人类使用的医疗器械。
对于本文提到的有粉手套,早在2016年3月21日,FDA公布一项提议(部分截图如下),拟在全美禁用医用有粉手套,该提议经过90天的公众评议后,2016年12月16日正式发布,并在2017年1月18日生效。
(二)哪些有粉手套被纳入了FDA的禁令?
A. 禁令清单中的有粉手套
2017年1月18日起,FDA在生效有粉手套的禁令时,同时修改了其联邦法规,将有粉外科手套、有粉患者检查手套、用于润滑外科手套的可吸收粉末纳入禁止器械的清单,清单中具体给出的定义如下:
Ø 依据21 CFR 895.102 ,有粉外科手套是指一种用于在手术室人员手上佩戴的设备,用于保护手术伤口免受污染。有粉外科医生的手套将粉末用于生产以外的目的。
Ø 依据21 CFR 895.103, 有粉患者检查手套是指用于医疗目的的一次性设备,佩戴在检查人员的手或手指上,以防止患者和检查人员之间的污染。有粉患者检查手套将粉末用于制生产以外的目的。
Ø 依据21 CFR 895.104,用于润滑外科手套的可吸收粉末是指是由玉米淀粉制成的粉末,符合美国药典(U.S.P)中可吸收粉末的规格,用于在戴上外科手套之前润滑外科医生的手。该设备可通过生物降解吸收。
B. 除外的情况
不过需要特别说明的是,有两类情况需要注意是排除在有粉手套的禁令外的:
1)有粉辐射保护手套
在FDA禁止的有粉手套中,对于有粉辐射保护手套是不包含在内,因为按照FDA的描述,目前不知道市场上有任何有粉辐射保护手套,且FDA缺乏证据来确定是否满足该类手套的禁止标准。因此,在这次的禁令中,有粉辐射保护手套是排除在外的。
2)在生产过程中使用粉的无粉手套
此次的禁令提到是禁止有粉手套,但我们发现在21 CFR 895的禁令清单中提及是指“有粉将粉末用于生产以外的目的”,就是说将粉末用于生产是可以的,且在禁令文中也提及不适用于无粉手套制造过程中使用的粉末,因为该粉末不属于最终成品手套的一部分。不过对于成品无粉手套中的残留粉末给出了一定的限度要求,建议如果预计在生产工艺中使用粉末,需要确保成品手套中的粉末尽可能少,在2008年FDA发布的《医用手套指导手册》中建议依据ASTM D6124A标准测定,无粉手套每个手套的残留粉末和碎屑不超过2毫克。
(三)有粉手套为什么会被禁止呢?
在有粉手套发布禁令中对被禁的原因有进行描述,是由于这些手套对医生和患者依然构成“不合理、较大的染病或伤害风险”,这些风险无法通过使用新标签或更新标签来消除。这些风险主要在于当内部身体组织暴露在粉末中时,患者和卫生保健提供者会面临包括严重的气道炎症和过敏反应,粉末颗粒也可能触发身体的免疫反应,导致组织在颗粒(粒细胞瘤)或疤痕组织形成(粘附)周围形成,这可能导致手术并发症。关于这项禁令,FDA CDRH 中心主任Jeffrey Shuren提到:"这项禁令是为了保护患者和卫生保健专业人员免受他们可能甚至不知道的危险。” "我们非常严肃地对待禁令,只有在我们认为有必要保护公众健康时才采取这一行动。”
(四)现在哪些手套可以替代使用呢?
FDA发布2016年禁令的同时,除将禁止有粉手套纳入禁止器械的清单中,也充分评估可替代的无粉手套的情况,并对相关的分类法规进行修订,目前替代有粉手套的产品在FDA的监管主要如下2大类:
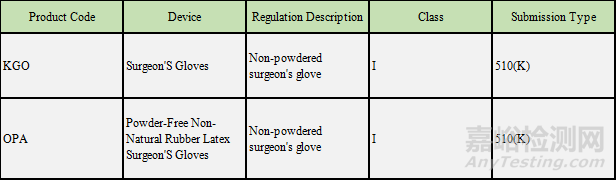
一类是监管联邦法规21 CFR 878.4460对应的无粉外科手套,主要的产品代码如下:
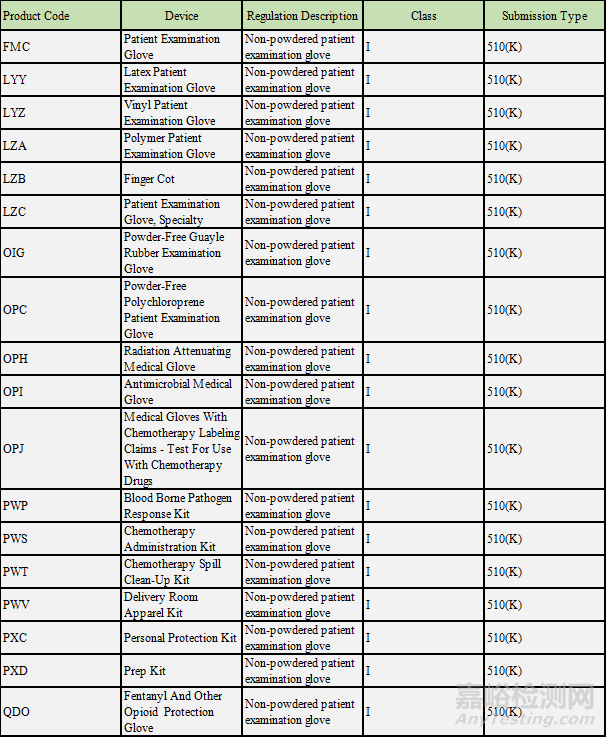
还有一类是监管联邦法规21 CFR 880.6250对应的无粉病人检测手套,主要的产品代码如下:
在FDA对于外科手套和患者检查手套是分为I类,上市类型是510(K),由于这次禁令将原来的有粉手套从产品代码中进行删除,且在现有产品代码的设备描述中明确标示无粉手套,这就意味着医用有粉手套可能无论如何无法获得美国FDA的批文。
(五)除有粉手套外,历史上FDA还颁布过哪些禁令呢?
虽然FDA有权利依据《联邦食品、药品和化妆品法》第516(a)节的要求禁止医疗器械,但据FDA CDRH信息来源,FDA很少对这一权力采取行动,截止目前可查询的发布的禁令共3项,清单如下,即除了上面提及的有粉手套外,还包括假发纤维和用于自残或攻击行为的电刺激设备。在这里呢,一并给大家普及了解一下。
|
设备名称 |
禁令生效日期 |
|
用于自残或攻击行为的电刺激设备 (ESDs) |
2020年4月6日 |
|
有粉外科手套、有粉患者检查手套、用于润滑外科手套的可吸收粉末 |
2017年1月18日 |
|
假发纤维 |
1983年6月3日 |
1.假发纤维
FDA最早发布禁止的器械是1983年6月3日生效的假发纤维,在查询到的关于FDA禁止的原因中提到,FDA发现这种假发纤维对公众健康没有好处,且关于这种器械的好处上对患者或用户造成重大欺骗,认为假发纤维既不刺激头发生长,也不遮盖秃顶,但实际上可能导致严重的感染、疾病和植入损伤,而且FDA认为,通过标签和宣传材料会存在直接或暗示歪曲设备的安全,有效,并造成很少或没有不适,以及其他误导性的说法。
关于假发纤维禁止器械在FDA 21 CFR 895.101中给到定义,具体指的是:假发纤维是用于植入人类头皮以模拟自然头发或隐藏秃顶的器械。假发纤维可能由各种材料组成:例如,合成纤维,如模态纤维、多晶硅纤维和聚酯纤维,以及天然纤维,如加工过的人类头发。不过有一种情况是排除在禁止之外的-自然头发移植,这种情况是指其中一个人的头发及其周围的组织被手术从人头皮上的一个位置切除,然后移植到这个人头皮的另一个区域。
2.用于自残或攻击行为的电刺激设备
2020年3月6日,FDA公布了一则关于禁止使用进行自我伤害或攻击行为的电刺激装置(ESDs)的联邦公告,因为该设备存在不合理和实质性的疾病或伤害风险,无法通过标签来纠正或消除。此禁令包括已分发和使用的新设备和设备,该禁令于2020.4.6生效。
ESDs是一种可逆的调节设备,可对一个人的皮肤施加有害的电刺激(冲击),以减少或停止此类行为。SIB (自残行为)和 AB(攻击行为) 经常同时表现在同一个人身上,智力或发育障碍患者表现出这些行为的比率很多。SIB通常包括头部撞击、手咬、过度划伤和割皮,更极端可能会导致出血、骨折甚至因挖眼、戳眼而失明、其他永久性组织损伤、吞咽危险物体或物质造成的伤害等。AB 涉及反复的人身攻击,可能对个人、他人或财产造成危险。
在这次FDA禁止的电刺激设备(ESDs)是指通过连接到个人皮肤的电极进行电击,以试图使他们停止从事自残或攻击行为。在其中提到许多接触到这些设备的患者有智力或发育障碍,使得很难表达他们的痛苦或同意,这意味着许多表现出SIB或AB的人是弱势群体的一部分,从而会引申出使用这些设备会带来相关的一些重大心理和身体风险,包括抑郁、焦虑、自我伤害行为恶化以及创伤后应激障碍、疼痛、烧伤和组织损伤的症状。此外,设备故障还存在错误电击的风险。由于这些风险无法通过新的或更新的标签来消除,因此必须禁止该产品以保护公众健康。
需要特别说明的是:受此禁令约束的ESD是旨在减少或停止SIB或AB的逆向调节设备。逆向调节是将有毒刺激(如ESD对个人皮肤的有害电击)与目标行为配对,使个人开始将有害刺激与行为联系起来。预期的结果是个人停止从事行为,并随着时间的推移,成为条件而不表现目标行为。有些ESD用于其他目的,如戒烟等,但是禁令只包括那些旨在减少或消除SIB或AB的设备。ESD不用于电惊厥治疗,有时称为电击疗法或ECT,这些与这一规则是无关的。
关于该禁令提到的器械在21 CFR 895.105中进行了定义,具体是指:用于自残或攻击性行为的电刺激设备是逆向调节设备,用于对人的皮肤施加有害的电刺激,以减少或停止自我伤害或攻击行为。
(六)FDA是如何发布医疗器械禁令的?
前面提到了FDA发布的3项禁令,那FDA发布禁令的流程是怎么的呢?具体如下:
当FDA决定禁止某器械时,是通过分析和权衡器械对个人构成的风险和好处,这些分析可能包括:
· 识别和研究器械,包括评估不良事件;
· 分析当前医疗实践中使用的替代设备和治疗方法带来的风险和益处;
· 分析设备上标签的更改是否降低了风险;
· 评估医学文献;
· 与外部专家举行小组会议;
· 与专业协会讨论相关事项;
· 评估来自保健专业人员和患者的信息。
FDA 可以禁止没有实际疾病或伤害证据的设备,并且只需要发现设备有可能根据所有可用的数据和信息有潜在呈现的风险程度。
FDA可能会发起器械的禁止,如果:
· 该设备在标签上存在重大欺骗性或不合理且实质性的疾病或伤害风险,以及
· 这种欺骗或风险不能或尚未通过标签或标签更改来纠正或消除。
如果FDA决定启动程序来禁止一种设备,其提议制定的规则将在联邦公报(Federal Register)上发布,禁令通常分为提议禁令(Proposed Ban)和终版禁令(Final Ban)两个阶段,对于提议禁令通常会有至少30天的评论期,不过在某些情况下,FDA可以在提议禁令中规定一个特别的生效日期,一旦禁令在联邦公报上公布,该禁令就会立即生效。当 FDA 确定所涉及的潜在或实际伤害足够严重, 以至于认为将危及已经或即将接触该设备的个人的健康时,可以使用此程序。
(七)结语
医疗器械的安全和有效作是监管的核心,对于被列入发布“ban”的医疗器械,不论对于生产企业、使用单位、产品用户来说,都是需要额外引起关注的,对于其他监管当局也是需要为考虑本国器械的安全有效需要引以借鉴的。

来源:Internet


