您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-02-20 23:58
摘 要 / Abstract
近两年制药行业接连从缬沙坦、雷尼替丁、二甲双胍等药物中发现亚硝胺类杂质残留,反映出原有的基因毒性杂质的评估与控制策略存在着一些问题和缺陷,表明制药工业界和监管部门对亚硝胺基因毒性杂质的认知需要一定的发展和提升空间。如何避免亚硝胺类杂质残留事件的发生,对于制药工业界和监管部门都是一个巨大的挑战。本文简要的回顾了基因毒性杂质的历史和现状,对各国监管部门的应对策略进行解读,并对缬沙坦、雷尼替丁、二甲双胍等药物中产生亚硝胺类杂质的机制进行探讨。笔者结合文献资料、ICH 指导文件以及近期监管部门发布的相关指导原则,从基因毒性杂质识别、原料药和成品工艺风险评估、毒理学评估、控制策略、分析方法等方面进行阐述,以期助力基因毒性杂质研究的进一步发展。
The presence of nitrosamine impurities in valsartan, ranitidine, metformin and other drugs has been reported one after another in the past 2 years, which reveals problems with the original risk assessment and control strategy of genotoxic impurities. These events also indicate that it takes time and a progressive process for both the pharmaceutical industry and the regulators to have a profound and complete understanding of nitrosamine impurities. It is a great challenge, facing the industry as well as the regulators, to prevent these events from happening. This paper briefly reviews the history and current situation of genotoxic impurity studies, examines control and mitigation strategies by regulators of different countries/regions, and discusses the plausible mechanisms for nitrosamine formation in sartans, ranitidine and metformin. Based on literature, ICH guidance and relevant guidelines recently issued by regulators, the authors elaborate on the identification and evaluation of genotoxic impurities,risk assessment of drug substance and finished product processes, toxicological assessment, process control strategy and analytical methods in an effort to formulate an effective and readily implementable control strategy for genotoxic impurities.
关键词 / Key words
基因毒性杂质;ICH ;亚硝胺;风险评估;控制策略;分析方法
genotoxic impurities; ICH; nitrosoamine; risk assessment; control strategy;analytical method
基因毒性杂质(genotoxic impurities,GTI)又称遗传毒性杂质,是指化合物本身能直接或间接损伤细胞DNA,产生基因突变或体内诱变,具有致癌可能或者倾向。研究基因毒性杂质的根本目的是控制和降低其在人体中可能引发癌症的风险。致癌物按作用机制可分为基因毒性致癌物与非基因毒性致癌物,药物研发与生产中遇到的致癌物绝大部分是基因毒性致癌物。基因毒性致癌物的主要作用机制是造成DNA 损伤,而非基因毒性致癌物的作用机制比较复杂,包括氧化应激、酶活性的调节、细胞增殖与细胞凋亡等。对于化学物质可能引发癌症的毒性,可分为基因毒性、致突变性和致癌性3 个层面。绝大部分致癌物和诱变剂都具有基因毒性,但并非所有的基因毒性杂质都具有致突变性或致癌性。
药物的基因毒性杂质与普通杂质研究的前期发展历程相似,两类杂质均是从20 世纪80 年代末~90 年代初开始系统研究。推测其原因可能是由于随着药物分析的不断发展,更灵敏的仪器如高效液相色谱(HPLC)等开始被大规模地应用于药物分析和检测,因此一些药物杂质(包括基因毒性杂质)逐渐被检测出。同时,随着对基因毒性杂质认知的不断发展,相关的监管指南也在不断更新。近几年,制药行业接连从沙坦类、雷尼替丁、二甲双胍等药物中发现亚硝胺类杂质残留[1]。如何避免亚硝胺类杂质残留事件的发生,对制药工业界和监管部门都是一个巨大的挑战。
1、基因毒性杂质的定义、性质与分类
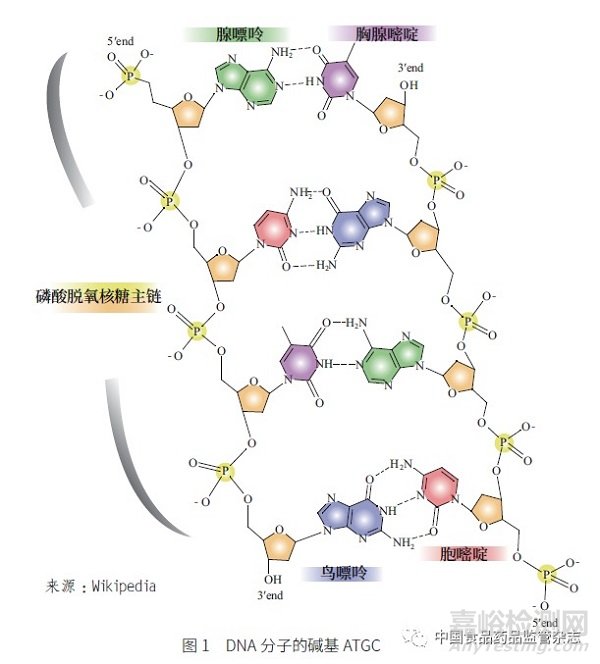
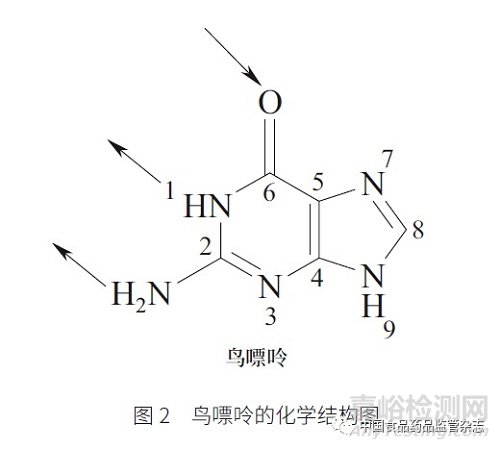
在药物研发的实际工作中遇到的基因毒性杂质, 绝大部分是烷基化试剂(alkylating agents),其化学基础为亲电性,即其本身是亲电试剂(electrophiles)。这是由于双螺旋DNA 分子含有4 种碱基,分别是腺嘌呤(adenine,A)、胸腺嘧啶(thymine,T)、鸟嘌呤(guanine,G)和胞嘧啶(cytosine,C),见图1。这4 种碱基均为亲核试剂(nucleophiles),因此亲电的烷基化试剂能够和亲核的DNA碱基发生化学反应。如鸟嘌呤(图2)发生烷基化的位点主要在O-6 位和N-7 位,其中O-6 位点的烷基化比较容易诱导基因毒性的产生。烷基化试剂可分为直接烷基化和间接烷基化试剂2 种。通常亲电性越强,其基因毒性越强,特别是需要经过代谢激活产生的基因毒性杂质(如间接烷基化试剂)。然而,对于直接烷基化试剂,当其反应活性太活泼时,其基因毒性反而可能减小。这可能是因为这类物质在还未进入细胞或未达到DNA 作用靶标时就已在体内分解,而在体内分解时所发生的化学反应也可能导致这类物质产生其他毒性。
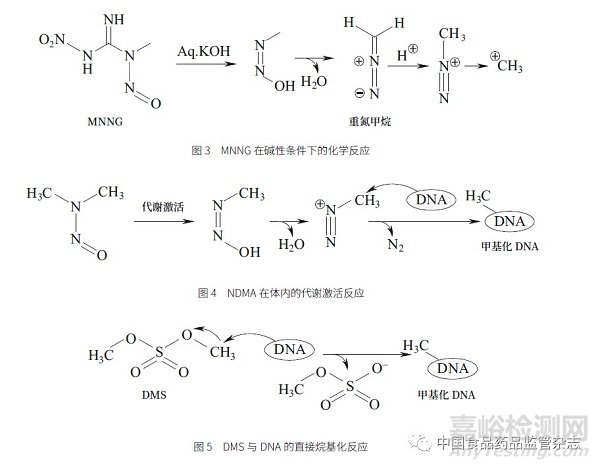
1968 年Singer 等[2] 首次报道了亚硝基胍的化学反应,1- 甲基-3- 硝基-1- 亚硝基胍(MNNG)在碱性条件下分解,经脱水反应生成重氮甲烷,重氮甲烷本身也是一个很强的烷基化试剂,进一步反应形成更加活泼的烷基化试剂,即甲基正离子(图3)。与亚硝基胍的化学反应相似,缬沙坦中发现的N- 亚硝基二甲胺(NDMA)杂质,通过代谢激活,也产生了同样的中间体,经脱水反应产生重氮甲烷,进一步反应生成甲基正离子。这2 种烷基化试剂均能与DNA 发生烷基化反应,生成甲基化DNA(图4)。直接烷基化试剂如硫酸二甲酯(dimethyl sulfate,DMS),可与DNA 一步反应生成甲基化DNA( 图5)。
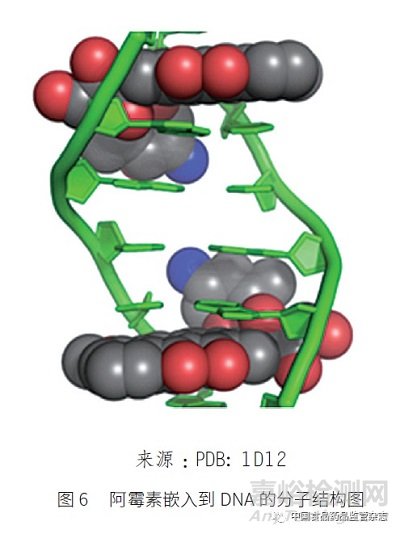
除了烷基化试剂外, 其他类型的基因毒性物质:如DNA 嵌入剂(DNA intercalator)阿霉素,是由阿霉素分子的平面部分嵌入到DNA 双螺旋结构(图6);重金属离子(heavy metal ions)可以与DNA 双螺旋的碱基键合;辐射(radioactivity)能造成DNA 链的断裂和交联,使DNA 遗传特性发生改变,产生基因毒性。
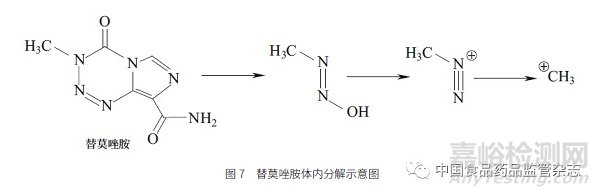
在靶向药物出现以前,有相当一部分化疗药物(包括大部分抗癌药)实际上是具有基因毒性的药物。如治疗脑癌的一线用药替莫唑胺(temozolomide,图7),能通过血脑屏障,最终分解形成甲基正离子。由此可知,基因毒性药物具有两面性。此类基因毒性药物对正常细胞和癌细胞没有区分能力,对人体快速生长的细胞组织(如毛发)同样也有抑制作用,所以接受这类传统化学药物治疗的癌症患者,通常会有脱发的现象,而通过靶向药物治疗的患者不良反应中一般没有脱发。
2、基因毒性杂质研究与监管简史
(一)基因毒性杂质相关的指南
1997 年首次发布的ICH Q3C《杂质:残留溶剂的指导原则》(Impurities:Guideline for Residual Solvents)中对具有遗传毒性的溶剂进行了限度规定;2006年1 月FDA 发布了行业和评审人员指南《推荐的遗传毒性试验结果综合分析法指导原则》(Guidance for Industry and Review Staff: Recommended Approachesto Integration of Genetic Toxicology Study Results),有关于遗传毒性研究数据整合的推荐方法;2006 年欧洲药品管理局(European Medicines Agency,EMA)人用药品委员会(CHMP)发布了《基因毒性杂质限度指南》(Guideline on the Limits of Genotoxic Impurities);2014 年首次发布的ICH M7《评估和控制药物中的DNA 活性(致突变)杂质以限制潜在的致癌风险》[Assessment and Controlof DNA Reactive (Mutagenic) Impuritiesin Pharmaceuticals to Limit Potential Carcinogenic Risk] 是协调了各方意见后发布的第1 个基因毒性杂质研究和控制的指南,其标题本身便指明了该指南的目的是评估和控制药物中具有DNA 反应活性(致突变)的杂质,以限制其在人体中的潜在致癌风险;2018 年3 月更新了该指南,最新版为ICH M7(R1)。此外, 在ICH M7(R1) 的附录中, 列出了14 个常见基因毒性杂质的可接受摄入量(acceptable intakes,AIs), 如4- 氯苯胺的终生可接受摄入量(lifetime AI)为34 μg/d ;氯乙烷的终生可接受摄入量为 1810 μg/d。需要指出的是,这些限度高于根据毒理学关注阈值(threshold oftoxicological concern,TTC)原则设定的、对于没有特定毒理学数据的潜在基因毒性杂质的限度(1.5 μg/d);特别是氯乙烷的限度比普通杂质的限度要高很多,在国际癌症研究机构(International Agencyfor Research on Cancer,IARC) 的分类中将其归类为3 类化合物,即目前没有足够证据表明能在人体导致癌症的化合物(需要特别注意,由于IARC 的分类与ICHM7 的分类及含义不同,因此不能混淆)
而在亚硝胺类杂质残留事件发生后,各国监管部门出台了一系列对亚硝胺风险评估和控制的指导文件[3-6]。
(二)警示结构
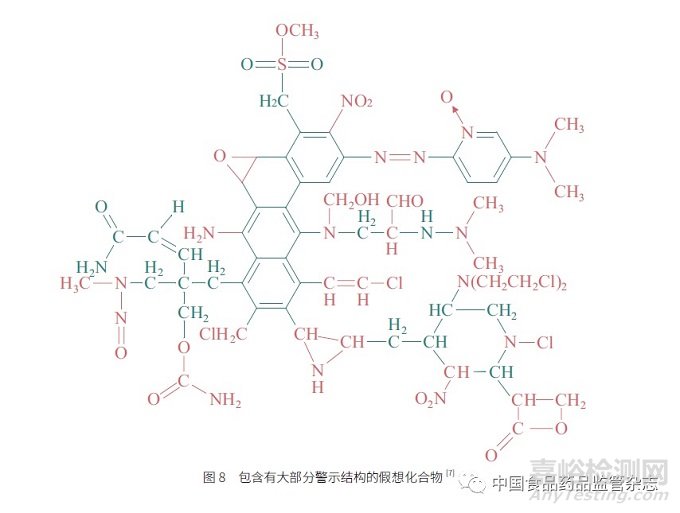
基因毒性杂质的风险评估,首先需要关注被评估的化合物是否具有警示结构。警示结构有很多种,其中绝大部分是亲电基团或经过代谢激活后可以转换成亲电基团的结构。1988 年Ashby 等[7] 假设了一个超级化合物,将很多警示结构包括在此假设化合物中(图8)。而具有警示结构的化合物并不一定具有基因毒性,只是其具有基因毒性的可能性较一般化合物高,但如果不对具有警示结构的化合物做进一步的毒理学评估,它们即是潜在的基因毒性杂质(potential genotoxic impurity,PGI),需要作为基因毒性杂质(genotoxicimpurity,GI)来控制,因此有时这2 个术语可能被混用。随着对于警示结构与基因毒性杂质认知的不断深入,这个超级化合物中包含的警示结构需要不断更新,原有的一些警示结构可能由于当时的数据局限或对数据分析的不完整,会被不恰当地归入。如吡啶氮氧化物,近期研究显示其引起基因毒性的可能性很小[8],但是由于被不恰当地归入警示结构,在申报人提交新药申报申请时,不论是国内还是国外,在涉及吡啶或吡啶类化合物时,审评员经常会要求申报人对此类氮氧化物形成的可能性进行考察,并要求评估其潜在基因毒性风险。而吡啶及吡啶类基团本身是缺电子基团,是一种弱亲核试剂,而形成吡啶氮氧化物的绝大部分过程是亲核氧化过程,所以作为降解物,吡啶及吡啶类的氮氧化物一般不易在制剂产品特别是固体制剂产品中形成。
( 三) 属于关注队列(cohort ofconcern)的警示结构
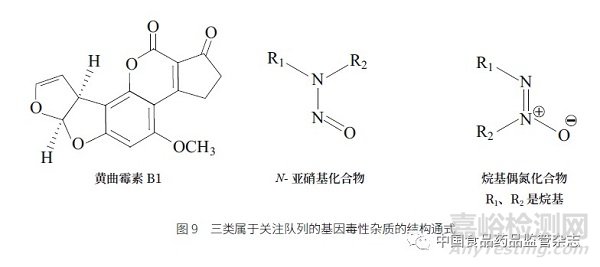
黄曲霉素类、N- 亚硝基和烷基偶氮氧化物这3 类杂质,经过代谢激活后基因毒性非常强,因此是需要研究者高度关注的3 类化合物,结构通式见图9。黄曲霉素通常在肝药酶的作用下,二氢呋喃环的C=C 形成非常活泼的环氧环,遇到肝细胞DNA 会导致DNA 烷基化。黄曲霉中毒性最强的是黄曲霉素B1(图10)[9],如果摄入黄曲霉素超过一定限量,很容易引发肝癌。
3、基因毒性杂质的风险评估
国家药品监督管理局(NMPA) 在2020 年5 月发布了《化学药物中亚硝胺类杂质研究技术指导原则(试行)》[6] 指导纲领,特别提到了“风险评估方法可以采用ICH Q9(《质量风险管理》)中所述的FMEA(Failure Mode Effects Analysis)或FMECA(Failure Mode,Effects and Criticality Analysis), 或其他科学合理的方法”。但在实际操作中,FMEA 和FMECA 这2 种模式在使用和应用上,特别是对是否存在亚硝胺类杂质进行的风险评估较为繁琐。
在实际操作中,对于原料药的风险评估,一般从几方面入手:①要从理论分析,分析其制造工艺是否有存在或产生基因毒性杂质的可能性(包括工艺杂质与降解杂质)。②要看基因毒性杂质产生的可能性及其残留可能性的大小。③要看基因毒性杂质的产生点到成品化学原料药(API)之间有几个步骤,如分离或纯化步骤,评估其去除率(purging factor)。
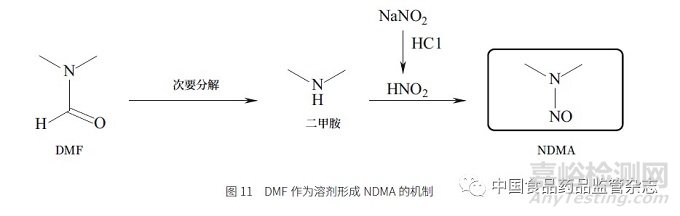
在过去的基因毒性评估中,风险评估控制点通常是评估起始物料、中间体、试剂、溶剂本身是否具有警示结构,以及评估其在反应中形成基因毒性杂质的可能性。2018 年7 月“缬沙坦事件”,即从缬沙坦原料药中检出NDMA 以来,提示着我们还必须关注物料中的杂质或是降解杂质是否具有警示结构,以及其在反应中是否有形成警示结构的可能性,如在“缬沙坦事件”中部分原料药工艺中使用二甲基甲酰胺(DMF)作为反应溶剂产生NDMA的机制(图11)。
在我们的实践中,形成了评估亚硝胺杂质风险行之有效的4 因素法(4 factoranalysis),即风险评估要关注以下4 个因素:①评估反应中是否用到仲胺。②评估反应中是否存在过亚硝酸(即亚硝酸盐与酸性反应条件)。③评估仲胺与亚硝酸是否在同一个反应步骤。④评估亚硝胺类杂质的潜在产生点之后,有几步分离或纯化步骤(即去除率是多少)。
前3 个因素为导致亚硝胺产生的风险点,而第4 个因素是降低风险的去除率。如果前3 个评估结果都是“有”或“是”的话,那么这个工艺产生亚硝胺的风险很高,即使有多步分离或纯化步骤,可能也无法完全控制住风险。相反,如果前3 个评估结果都是“无”或“否”的话,那么这个工艺产生亚硝胺的风险就很低或可忽略不计。从控制策略层面,首选的工艺最好能符合后者的情形,即以不产生亚硝胺类杂质的工艺为最佳、以去除纯化为辅的策略。
对于上述4 个因素,需要关注相应的细节。如果仲胺本身不是试剂,而是试剂中的杂质,其风险由于含量比之试剂的用量降低而相应降低。这是因为仲胺与亚硝酸的反应是直接形成亚硝胺的二级反应,其反应速率与仲胺和亚硝酸的浓度成正比。同样,叔胺的风险比仲胺低很多,因其反应形成亚硝胺需要多个步骤,故效率会降低很多。根据我们以三乙胺和二乙胺为模型在不同浓度范围内进行的初步研究,三乙胺与亚硝酸形成N-亚硝基二乙胺(NDEA)的效率只是二乙胺的1/50~1/200。上述4 因素也可以量化,根据每一类工艺的特点,赋予4 个因素(F1、F2、F3、F4)以合理的数值,以前3 个因素为乘积,除以第4 个因素而得到每一个工艺量化后的风险值,即风险=F1×F2×F3 ∕ F4,数值越低,则风险越低。
对于制剂中基因毒性杂质的风险评估,重点在于判断原料药中的基因毒性杂质是工艺杂质还是降解杂质,包括原料药与辅料相互作用形成的降解杂质[10],以及辅料和包材中可能引入的基因毒性杂质。对于液体和半固体制剂,尤其需要关注主包材中可能引入的基因毒性杂质。
4、基因毒性杂质的控制策略
对于原料药中亚硝胺类杂质的质量控制,需要注意避免高风险的反应、试剂、溶剂等,包括:①避免高风险的反应,如亚硝酸淬灭(在产品存在的情况下)、次氯酸钠淬灭(在产品存在的情况下)、重氮化反应(sandmeyer reaction)等。②避免高风险的试剂,仲胺或含仲胺杂质的试剂(如二乙胺、三乙胺等)。③慎用高风险的溶剂,DMF、N- 甲基-2- 吡咯烷酮(NMP)。④避免在工艺后期步骤采用一锅法,尤其是可能含有基因毒性或潜在基因毒性杂质的步骤。在充分理解基因毒性杂质产生的源头、走向、去除率(以加标实验估算去除率)的基础上,制定合理的控制策略及指标。
对于制剂中基因毒性杂质的控制,如果是原料药的工艺杂质,可在原料药中制定适当的指标控制;如果是降解杂质,需要在稳定性和加速稳定性研究时加以关注。通常温度和湿度会加速药物的降解,而氧气则会加速氧化降解杂质的产生;制剂产品的湿度和含氧量,可以通过选择相关的包材、干燥剂和除氧剂加以控制。
5、基因毒性杂质的毒理学评估
如果化合物具有警示结构且没有相关的毒理学研究数据,可以采用计算机软件进行评估(in silico evaluation),即利用符合经济合作与发展组织(Organisation for Economic Co-operation and Development,OECD) 要求的定量结构活性关系(Q)SAR(Quantitative structure activity relationship) 模型对化合物在细菌回复突变试验(Ames test)呈阳性的概率进行预测。ICH M7 指出“计算机毒理学应采用(定量)构- 效关系(Q)SAR 方法学进行毒性评估”“应采用两种互补的(Q)SAR 预测方法。一种方法应基于专家知识规则,另一种方法应基于统计学”。常用的评估软件有Derek/Sarah、Case Ultra(by MultiCase)、LeadScope、ACD/Labs 等。需要注意因为Derek 只是识别人类专家经验规则定义的警示结构的模型软件,而Sarah 只是基于统计学分析的(Q)SAR 模型的预测软件,Derek 和Sarah 软件需要同时使用。其他各家软件中均同时包含了这2 个预测模型功能。
化合物的毒理学数据来源包括ICHM7 附录、致癌性数据库(Carcinogenicity Potency Database,CPDB, 已被整合到其他数据库中)、欧盟化学品管理局(European Chemical Agency,ECHA)、国际癌症研究署(International Agencyfor Research on Cancer,IARC) 分类、美国国家癌症研究所/ 国家毒理学计划(NCI/NTP) 以及其他公开发表的文献来源等。CPDB 数据库提供了1547 个化合物的半数致癌剂量(TD50),可根据TD50数据线性外推出AI。IARC 分类定义包括1 类:已知的人类致癌物(已证实的致癌物或对人体有一定致癌作用的化合物);2A 类:可能致癌物(充分的动物致癌性研究数据,但人类致癌性研究数据有限);2B 类:可能致癌物(有限的人类致癌性研究数据,不充分的动物致癌性研究数据);3 类:对于人类的致癌性,无法归类的物质(不充分的人类和动物致癌性研究数据);4 类:不可归类为人类致癌物(充分的人类非致癌性研究数据)。
(一)数据库未更新数据的问题
常见的基因毒性杂质有磺酸酯、硫酸酯、苯胺类化合物、硝基化合物、氨基甲酸乙酯等。在实际审评时,可能会遇到一些特殊情况,如氨基甲酸乙酯在CPDB 数据库里的限度值远高于FDA 和EMA 采用的限度值,归其原因可能是数据库里的数据未及时更新。而监管部门在审核时,有的机构本身会有原研数据,因此对类似的引用数据库中未更新的数据,可能会不予接受。
(二)基于基团亲电性的经验规则
1. 含醛基化合物的邻位效应
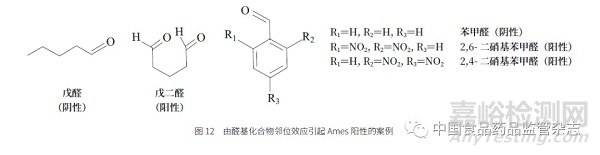
一些简单的含醛基化合物产生基因毒性的可能性比较小,而在醛基邻位含有吸电子基团的醛基化合物,其基因毒性的可能性增大。归其原因是邻位吸电子基团使得醛基化合物的亲电性变强,而这类化合物在反相高效液相色谱(RPHPLC)的分析中很可能以水合物的形式存在,这是醛基被活化的直接证据。另外,一些双醛基化合物可能由于2 个醛基的协同效应而产生基因毒性。一些此类案例如图12 所示,戊醛Ames 为阴性,而戊二醛Ames 为阳性;苯甲醛Ames为阴性,而2,4- 二硝基苯甲醛和2,6- 二硝基苯甲醛Ames 为阳性。
2. 含卤素烷基化合物
简单的含氯烷基化合物(即不与一些其他官能团相连接,如苄基、烯丙基等激活基团)是基因毒性杂质的可能性比较小或其基因毒性很弱,属于后者的化合物的AI 比常规杂质的限度高很多。可根据其亲电化学反应活性进行判断,如氯乙烷AI 为1810 μg/d、氯甲烷 AI 为1361 μg/d,二者都是很弱的亲电试剂。而简单的含溴烷基化合物是基因毒性杂质的可能性比同类含氯烷基化合物大很多,如溴乙烷的AI 为149 μg/d ;归其原因可能是其亲电性增加所致。
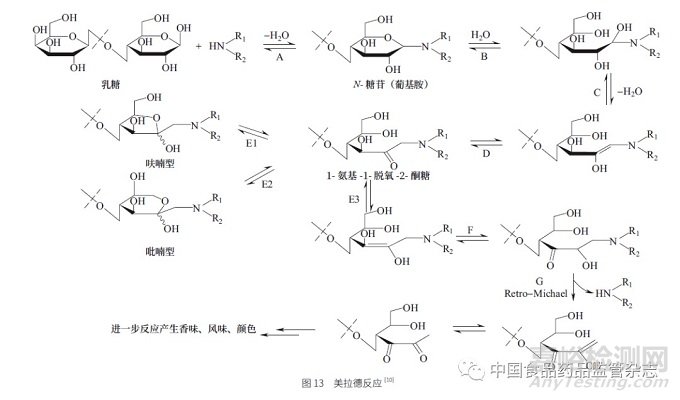
(三)警示结构杂质在食品中的广泛存在——美拉德反应(Maillardreaction)
美拉德反应在食品化学中具有重要意义,如制备蛋糕或烤面包时的色泽或香味,是由于在反应中产生了很多微量含酮或醛基的物质所产生的。这类物质很多具有警示结构,尤其是其中的α- 羰基类化合物。美拉德反应的示意图见图13。
(四) Ames 试验
评估一个化合物是否为基因毒性杂质的Ames 试验,按照目前OECD 的标准, 通常采用5 个菌株(Salmonella typhimurium TA1535 ;TA1537 或TA97或TA97a ;TA98 ;TA100 ;E. Coli WP2uvrA 或E.coliWP2 uvrA/PKM101 或Salmonella typhimurium TA102) 进行检测,其中4 个为鼠伤寒沙门菌突变株,1 个为大肠杆菌突变株。而在实际审评中,如果参照的是比较老旧的文献资料,没有同时在上述5个菌株中做相关试验,即使Ames 检测结果为阴性,监管部门在审核时也可能会不予接受。
(五)亚硝胺类杂质TD50 数值与合理限度的制定问题
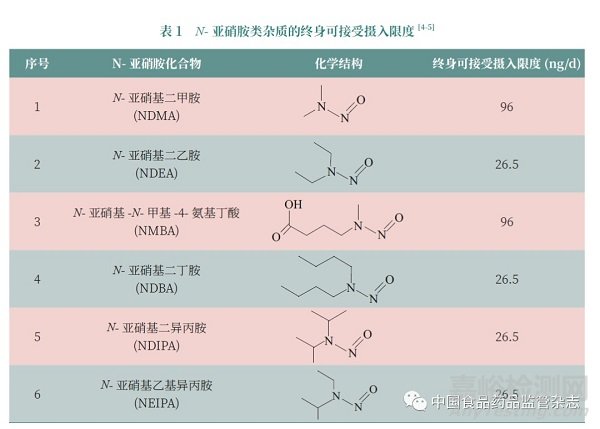
自“亚硝胺类事件”出现之后,从长远角度考虑,各研发机构均需采取有效的措施及制定合理的控制策略,以避免工艺中产生亚硝胺类杂质。各国监管部门对于亚硝胺类基因毒性杂质的控制策略有所不同,FDA 和EMA 在事件初期发布了亚硝胺类杂质临时限度(Interimlimits)[3]。随后,EMA 要求在2 年内对所有药物进行亚硝胺风险评估[11-13] ;FDA 要求在其2020 年9 月发布《人用药物中亚硝胺杂质控制》(Control of Nitrosamine Impurities in Human Drugs)指南后6 个月内,对已批准和上市的药物进行亚硝胺风险评估[4] ;NMPA 发布了《化学药物中亚硝胺类杂质研究技术指导原则(试行)》[6]。随着对亚硝胺类杂质认知的不断深入,监管部门接受在对亚硝胺类化合物进行充分风险评估的基础上,对致癌性研究数据充分的亚硝胺类化合物,可以采用TD50 进行线性外推得到其(终身)可接受摄入限度(acceptable intake limits), 如NDMA(AI = 96 ng/d)和NDEA(AI = 26.5 ng/d) 的可接受摄入量限度是根据其TD50 推算的(表1)[4-5]。而对于致癌性研究不充分的亚硝胺类化合物[ 如N- 亚硝基二丁胺(NDBA)、N-亚硝基-N- 甲基-4- 氨基丁酸(NMBA)和N- 亚硝基二异丙胺(NDIPA)], 即使其有TD50 值也不能直接采用,仍需要通过构效关系(SAR)分析制定合理限度;如NMBA 的可接受摄入量可以根据NDMA 的TD50 进行推算,NDBA 和NDIPA 的可接受摄入量可以根据NDEA的TD50 进行推算。对于致癌性数据未知的亚硝胺类化合物,EMA 推荐采用安全工作组(safety working party,SWP)的特定类别化合物的TTC 18 ng/d 作为保守限度;而对于结构类似的亚硝胺类化合物,也可以通过SAR 分析制定合理限度,如N- 亚硝基乙基异丙胺(NEIPA)的可接受摄入量可以根据NDEA 的TD50进行推算[5]。
6、基因毒性杂质的分析方法
基因毒性杂质分析方法的开发和选择不同于其他普通杂质分析方法,在灵敏度、选择性、待测物稳定性、基质复杂性等方面有其特殊要求。一般来说,基因毒性杂质的分析方法主要有限度方法和定量方法。如果原料药中的基因毒性杂质在至少6 个连续的中试批次或者3 个连续的生产批次中,测得结果均低于可接受标准的30%,则可以进行论述并定期检测,以后每年抽检3 批即可;如果不满足该条件,则需要对原料药进行常规批批检测。
(一)根据基因毒性杂质的限度和结构,开发合适的分析方法
基因毒性杂质检测通常采用的方法有:高效液相色谱法(HPLC)、液相色谱- 质谱联用(LC-MS)、液相色谱- 质谱/ 质谱联用(LC-MS/MS)、液相色谱- 高分辨质谱联用(LC-HRMS)、气相色谱法(GC)、气相色谱- 质谱联用(GC-MS)、气相色谱-质谱/ 质谱联用(GC-MS/MS,直接进样与顶空进样)、离子色谱法(IC)等。对于不同性质的基因毒性杂质,采取不同的方法:
挥发性杂质:采用GC、GC-MS 和GC-MS/MS 等检测方法。通常杂质限度在100 ppm 以上,可以直接采用GC 方法[ 氢火焰离子化检测器(FID)或电子捕获检测器(ECD)] ;若杂质限度在1~100 ppm,可以采用GC-MS 方法,通常采用电子电离(electron Ionization,EI) 模式;若杂质的限度低于1 ppm,推荐使用GCMS/MS 方法,以获取更好的灵敏度和专属性。
GC 方法需要考虑使用何种进样模式。顶空进样:适用于挥发性大的组分分析,可以减少样品的前处理步骤,减少样品分解对于目标杂质的干扰影响,优点是基质干扰小、进样重复性好;缺点在于由于杂质的分配系数原因导致基质对定量准确性可能产生一些影响。直接进样:适用于多数热稳定性好的组分分析,优点是能够在进样口气化的化合物,进样方式简单方便;缺点在于进样重复性差,并且由于大量样品进入进样口,长期检测会导致仪器污染的可能性大大增加。若样品制备时采用液液萃取(liquid-liquidextraction)的方法或采用 基质沉淀法,即先将样品溶解在良溶剂中,再用不良溶剂使样品析出,目标杂质仍处于溶液中,此法不但能提高方法的灵敏度,也可以减少样品对仪器的污染。
GC-MS 用于基因毒性杂质检测时,由于存在基质干扰,可能有共流出物质的碎片或者同位素峰刚好与目标杂质的表观质荷比(m/z)相同,这样就会导致检测结果的假阳性,特别是采集的m/z 较低的物质这种现象尤为突出。采用GC-MS/MS 可以降低假阳性的发生率,并可进一步通过观察样品与对照品检测的定性离子和定量离子的比例是否一致来帮助判断是否有假阳性。当出现假阳性时,需要通过优化GC 参数(进样口温度、柱温、色谱柱)将共流出物和目标杂质达到基线分离来解决假阳性,也可通过加入内标物(目标杂质的氘代物)进行校正。
非挥发性杂质:采用HPLC、LC-MS、LC-MS/MS 等检测方法。通常杂质的限度在100 ppm 以上,可以直接采用HPLC方法(取决于紫外或荧光发色团的强弱)。若杂质限度在1~100 ppm, 可以采用LC-MS 方法,通常采用ESI 正离子模式;但根据离子化基团的特性,ESI 负离子可能在某些情况下更合适;对于难离子化的化合物,通常采用APCI。若杂质的限度低于1 ppm,推荐使用LC-MS/MS 方法,以获取更好的灵敏度和专属性。同时,LC-MS/MS 方法可降低基质干扰造成的假阴性(离子抑制)和假阳性(共流出物或同位素干扰);对于无机阴离子和阳离子(如叠氮酸、亚硝酸、溴离子等)和有机离子(如生物胺、有机酸、糖类分析等)则可以采用IC 方法来检测。
(二)基因毒性杂质分析方法开发的难点
基因毒性杂质分析方法开发的难点在于样品基质对杂质的影响、杂质离子化效率、方法参数的选择等。
方法参数的选择:合理选择流动相(乙腈或甲醇),以确保目标杂质有合适的保留时间(k’),而不至于太靠近死体积,以避免可能受到的干扰。同时,有机相的类型和比例对离子化效率有较大影响。在质谱多反应监测(MRM)定量离子和定性离子的选择上,应选择丰度较高、受干扰较小的离子,减少同位素峰和共流出物的干扰,同时要考虑样品基质对离子选择的影响。
灵敏度的优化:在适合的仪器和与化合物性质匹配的离子源基础上,优化色谱和质谱参数;从化合物的结构入手,选择合适的稀释液和流动相,确保化合物能高效地离子化提高离子化效率;设计正交试验对于离子源的参数进行优化。同时,将仪器保持在最佳状态也是确保高效、稳定离子化的有效手段,否则,系统的残留或者污染既会对某些目标杂质产生吸附或者离子抑制作用,降低方法的灵敏度和重现性,也可能在有些场合导致假阳性结果。
对于一些质谱响应较弱的杂质,必要时也可采用衍生法,简单列举如下。
LC-MS 衍生:目的是引入易离子化基团(如2- 巯基吡啶、吡啶等)。卤代烷烃或含卤代烷基的杂质可以采用2- 巯基吡啶衍生;对于某些此类杂质,也可以用吡啶本身来衍生。前者引入的吡啶基团在酸性条件下,容易形成正离子;而吡啶直接衍生卤代烷基化合物所形成的N- 烷基吡啶正离子,在任何pH 范围内均为正离子,其质谱响应非常高,可明显提高检测方法的灵敏度。
GC-MS 衍生:目的是引入衍生基团使杂质变得更容易挥发,如全氟苯基类衍生试剂五氟溴苄(PFBBr)、五氟苯酚、五氟苯硫酚和碘化钾/ 碘化钠等。五氟溴苄可以衍生羧酸、酚类和磺胺类杂质;五氟苯酚、五氟苯硫酚可以衍生硫酸酯或卤代烃类杂质;碘化钾/ 碘化钠可以衍生磺酸酯类杂质,形成相应的碘代烷烃。这些衍生产物在GC-MS 上的响应均有明显提高,通常能解决方法灵敏度的问题。
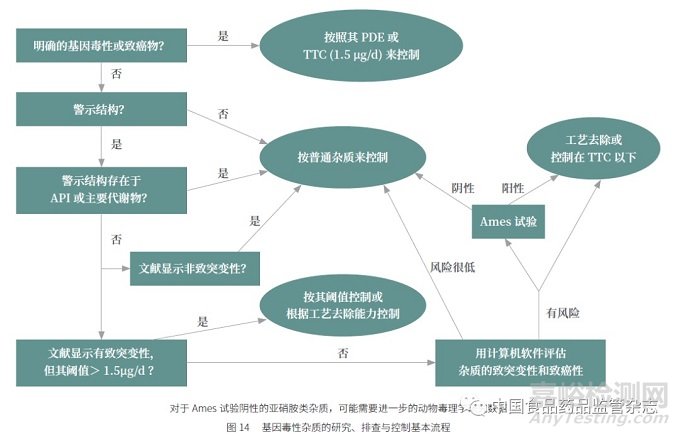
7、基因毒性杂质的研究、排查与控制基本流程
基因毒性杂质的研究、排查与控制基本流程见图14。有些案例可能需要结合文献和计算机软件评估的结果做出综合判断。
8、新挑战及未来趋势
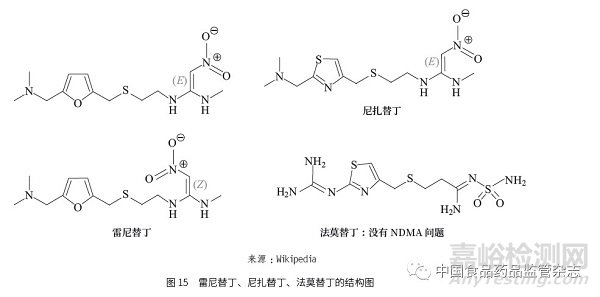
目前,制药行业和监管部门对基因毒性杂质还在不断的认知。这一点在FDA发布的相应公告中也有指出[1],其具体表现为亚硝胺类杂质在其他类型药物中不断被发现,如2019 年下半年发现雷尼替丁和尼扎替丁中有超出可接受限度的NDMA[14]。根据目前已知的信息判断,NDMA 是雷尼替丁和尼扎替丁的降解物。2016 年,美国的一项研究报道[15] 发现NDMA 也是雷尼替丁的代谢物。从雷尼替丁和尼扎替丁的结构判断(图15),这2个药物本身就具备了形成NDMA 降解物或代谢物的条件。结构中的硝基在热分解条件下可能重排成亚硝酸酯,进而有可能继续分解成亚硝酸,这样每分解1 个雷尼替丁和尼扎替丁分子就可能产生1 分子的亚硝酸,后者可以跟雷尼替丁和尼扎替丁结构中的二甲基胺基团发生反应而最终形成NDMA。从这个可能的机制看,其他替丁类药物如法莫替丁(图15),因为其结构中既没有硝基,也没有二甲基胺结构,因此不会形成NDMA。从临床角度看,可替代雷尼替丁和尼扎替丁的药物很多,除了其他替丁类药物,还有质子泵抑制剂类药物。
2019 年末在二甲双胍的部分缓释制剂中也发现了超出可接受限度的NDMA[16],二甲双胍目前没有同类替代药物。从目前得到的信息看,二甲双胍原料药和普通制剂中尚未发现有超标的NDMA,但是缓释制剂中存在降解杂质NDMA 的问题。根据我们的初步研究发现,NDMA 可能是二甲双胍的氧化降解杂质,用过氧化氢对二甲双胍溶液进行强降解,可随时间的不断增加而产生NDMA。
基因毒性杂质的风险评估、排查与控制,是一项跨部门、跨学科且具有相当挑战性的工作(图16)。随着我们对基因毒性杂质研究的不断深入,我们对其了解也将不断加深。监管部门对于基因毒性杂质限度的合理制定,也有着一个发展的过程。如在亚硝胺类杂质残留事件的最初发生阶段,FDA 的立场是亚硝胺类杂质不得存在(“Should be absent”);在这种情形下,亚硝胺的实际控制限度由相应检测方法的最低检测限(LOD)所决定。对于NDMA和NDEA,FDA 发布方法的LOD 分别为5和1 ppb(相对于缬沙坦的每日最大剂量而言)。随着事件的发生和发展,对于这2 个亚硝胺类杂质,FDA 现在接受由相应TD50 推算的AI, 分别为NDMA 96 ng/d和NDEA 26.5 ng/d(相当于在缬沙坦中的含量分别为300 和82.8 ppb)。另外,制定可接受限度(尤其是临时可接受限度)的一个重要考量是如果受到影响的药物召回,是否会造成这类药物的短缺,以及短缺所造成的后果是否会更严重。
笔者认为目前毒理学评估,尤其是对于Ames 阴性的亚硝胺类杂质,如何充分地评估、制定合理的限度仍不明朗;未来基因毒性杂质的风险评估、排查与控制将是原料药工艺开发的重中之重;基因毒性杂质分析方法的开发过程中,如何解决灵敏度、选择性、假阳性和假阴性问题也将是持续的挑战。

来源:中国食品药品监管杂志


