您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2020-02-12 16:25
由于无菌工艺对技术依赖性高并且对患者可能造成潜在的危害,无菌药物的生产工艺一直是药物生产过程中关键的工艺步骤,无菌生产过程工艺核查由各国药监部门分别执行,如美国FDA,WHO和EMA都要求对无菌工艺进行核查,因此知晓各个国家对“药品生产质量管理规范”(GMP)的规范条款以及条款适用情形对药企来说是十分必要的。
值得一提的是,大多数GMP规范条款仅描述了需要完成哪些工作项目,却没有叙述应该如何完成这些工作。因此这就需要企业建立起一套严谨审慎的系统方法来保证工艺符合GMP的规范要求。
本文对无菌药物相关的规范性指导原则和GMP基本的要求进行了概要性介绍,着重介绍工艺方面的规范要求。
生产工艺是产品质量保证的基础,与产品的质量控制同样非常重要。生产工艺规程必须事先确定且经过验证,生产过程必须严格执行经过验证的操作方法。无菌工艺操作要求人员间紧密配合协作,沟通交流,以及具备相应的设备体系,洁净室和相关设施,无菌组件。
本文列出了不同国家药监部门对厂房,实验室等级标准,区域等级标准,设备,人员,工艺和灭菌的要求(主要以WHO指导原则为主介绍,与其他地区指导原则对比的异同)
在设计和生产药物制剂工艺过程中,我们应该加强引入风险管理定义(ICH Q9)和现代制药质量体系(ICH Q10)的理念。无菌药物应该在洁净区域生产,根据工艺步骤我们可以将无菌制剂分为以下两类:
1. 终端灭菌的产品
2. 部分或者全部步骤实施无菌生产的产品
无菌制剂的生产
为了减少细微粒或者微生物污染给产品质量造成的潜在风险,无菌制剂每一步的生产操作环境必须具备规定水平的洁净度。
在产品的生产过程中,推荐使用单独的设备生产含活性微生物或者不含活性微生物的产品,除非产品含特定的灭活微生物、去活化/容器已经被充分的证明或者验证的情况除外。除此以外,在灭菌前的工艺过程也需要尽可能降低生物负荷。
WHO指导原则建议应该尽量缩短各个工序步骤之间的时间间隔,并且对间隔时间进行验证,例如,设备清洁和灭菌的间隔,设备灭菌和成型的间隔,成型和产品灭菌的间隔。除此以外,指导原则强调了隔离器技术(Isolator technology)和吹填封技术(blow-fill-seal technologies)的优势以及实施过程中的注意事项。隔离器和袖管或手套系统应该进行常规监测,以及依照特定频率进行泄漏测试是欧盟GMP和WHO重点关注内容。
环境监测计划是实验室控制的重要环节之一。监测计划应涵盖全部的生产班次,以及空气,地面,墙面和设备表面,尤其与产品,容器和包材直接接触的表面。
灭菌
通常来说,各个国家药监部门可以接受不同的灭菌方法,例如热灭菌,过滤除菌法,辐射灭菌法,环氧乙烷(EtO)灭菌;但是热灭菌法是首选使用的灭菌方法。各种负荷类型和负荷模式都需要经过验证。应该使用生物指示剂作为附加的灭菌监测方法,例如高压灭菌指示带和具有颜色变化的辐射指示片,都可以清晰的识别出物体处于的灭菌状态。
无菌药物应尽可能的选终端灭菌的方法,尽可能在最终容器中采用热灭菌方式进行。如果由于制剂的稳定性原因或者包装材料相容性的因素使产品不能采用最终容器热灭菌的方式,那么可以考虑采用过滤除菌的方法。由于无菌等级的滤膜无法完全去除病毒和支原体,这种情形下可以考虑采用一定程度的热灭菌方式作为过滤除菌的补充。
指导原则还建议在灌装前接近灌装点的位置采用另一新的无菌级别的滤膜进行二次过滤。滤膜的最大使用时间需要经过验证。滤膜的完整性测试也在指导原则中有所规定,滤膜纤维应该不脱落,并且滤膜的吸收或浸出过程应该不会影响产品组成成分。
FDA指导原则要求无菌测定方法应该是准确且可重复的;EU,GMP和WHO要求无菌制剂的测定方法应该是保证产品无菌的一系列质量控制的最终环节。测定应该使用的待测的产品进行验证。
人员
指导原则建议洁净区内的人员应该尽可能减少。在洁净区工作的人员,包括清洁工和设备维修工都应该接受培训,掌握生产(无菌技术),洁净室行为规范,人员卫生,穿着隔离服和微生物方面的基础知识。为了减少交叉污染,洁净室应雇佣称职人员,否则需要执行极其严格的清洁流程操作。
严格禁止佩戴手表,首饰,涂抹化妆品。指导原则还给出了隔离服适用的要求,包括洁净室等级,隔离服材料质量,更换衣服手套的频率,手套的消毒,独立专用的洗衣设备。
各个国家的药监部门一致认为,厂房或者区域的设计应该满足生产操作过程中微生物和微粒方面的相关要求,同时也要符合设备,组分材料和产品的要求。
建议采用光滑,防渗,易于消毒的表面,以及密封吊顶。A或B级区域建议避免安装水池。其他洁净级别的区域内下水管应配备带有空气阻断功能的装置,地漏应该带有水封/阻断功能。
在房间压差,空气流速,气流模式,更衣室连锁系统,更衣室数量(建议入口和出口安排单独的房间)。门应朝向高压侧打开,并且要配有自锁系统,以限制人员进出关键生产区域。
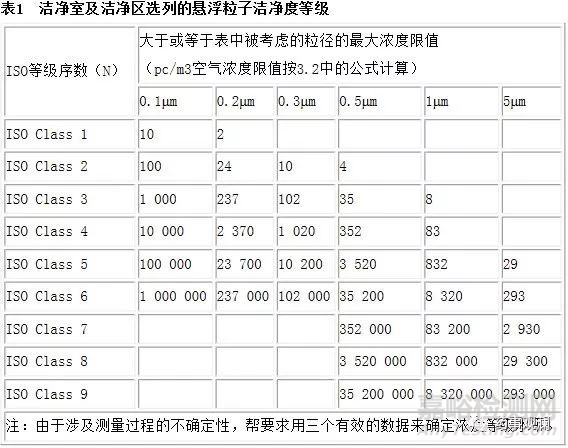
空气中颗粒物浓度的洁净等级划分应该依照ISO 14644-1标准中洁净室的分类标准(表1)。以下公式定义了最大允许粒子浓度,Cn (每立方米的粒子数量),粒径D(微米)
其中:
Cn 为大于或等于考察粒径的粒子的最大允许浓度(每立方米的粒子数量)
N为ISO 等级级别
D 为考察的粒径 (微米)
US FDA 规定仅考察运转的条件,粒径仅考察0.5um。EU GMP和WHO认为“静态”和“动态”条件的限度标准应该相同,5um和0.5um的粒径都需要进行考察。EU GMP和WHO指导原则没有定义D级的“动态”限度标准;制药企业应该综合风险分析结果和历史数据(如适用),建立企业内部“动态”限度标准。
动态生产过程中,生产厂应该对空气粒子和微生物污染的状态进行监控。FDA没有将培养皿法作为连续空气取样的强制方法。但是WHO/EUGMP 强制要求培养皿法和手套标识。
新版EU GMP 附件15(校验和验证)已经向公众发布,新版文件对设施设备,装置和流程的校验和验证进行介绍。附件15 还对比给出了比较其他法规的区别,包括EudraLex 4卷I部的其他内容,II部,附件11,ICH Q8,Q9,Q10,Q11,QWP 法规体系中关于工艺验证以及生产技术变更的内容指导原则。
设备
生产设备方面,指导原则建议传送带不能在A/B级洁净区域与较低级别的洁净区之间传送,除非传送带本身能够持续执行灭菌。此外,建议在洁净区域外放置安装设备;在设备维修后,应对设备和洁净室进行清洁和消毒/灭菌,除非维修时对洁净度和无菌状态没有影响时可以省略此步骤。WHO推荐应尽可能使用干热/湿热灭菌法来对设备进行灭菌。
其他指导原则
国际制药工程协会(ISPE)撰写的关于无菌药物生产设备的指南,在设施设计,建造,试运转和校验方面内容十分具有参考价值。该文件以本文引用的指导原则为基础,提出了大量在实践过程中的操作指导,以及实践良好操作规范过程中的大量丰富详实的案例。
除此以外还有一些其他行业标准和指导原则,如ISO 13408-1 standard“健康保健产品的无菌工艺”对于无菌工艺的设计非常有帮助。
小结
本文详实而系统地介绍了如何满足上述指导原则要求的方法,目的是帮助制药企业科学地管理产品的生产设施和工艺流程,从而符合严格规范的GMP技术要求。企业只有做好这些才能保证产品最终上市后,患者服用药物的安全性和有效性。
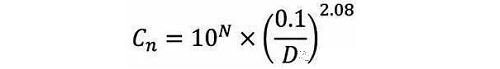

来源:药事纵横


