您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2024-09-18 12:08
前言
联合用药是指两种或两种以上的药物同时或先后联用,以增强疗效或降低毒性。临床前药代动力学实验中,通过动物体内联合给药的方式,改变相关代谢酶或者转运蛋白的活性,进而影响同服药物的机体内处置,在此过程中桥接体外和临床药代动力学相互作用研究。另一方面,基于药物相互作用的联合用药可以研究同服药物的体内处置机制,有助于同服药物的前期优化,从而得到新一代的药物。本篇文章就临床前药代动力学实验中常用的联合给药及其实验设计进行总结并举例说明。
1、与转运体抑制剂进行联合给药
药物转运体是一类位于细胞膜上的具有转运功能的蛋白质,广泛分布于机体内的各个组织,在药物的体内处置过程中发挥了重要作用。药物转运体包含两个不同的超家族,ABC转运蛋白(ATP-binding cassette transporters)主要为外排转运体,溶质转运体(solute carrier,SLC transporters)既有外排转运体也有摄取转运体。在不同的临床前动物种属中,转运体的表达水平,可能存在种属差异。如表1所示,P-糖蛋白(P-gp)和有机阴离子转运多肽(OATP1B1/1B3)在不同动物种属中表现出差异[1]。以下以P-gp和OATP1B1/1B3抑制剂联合给药为例展开讨论。
表1. P-gp、OATP1B1/1B3转运体在不同动物种属中的基因名称和mRNA表达水平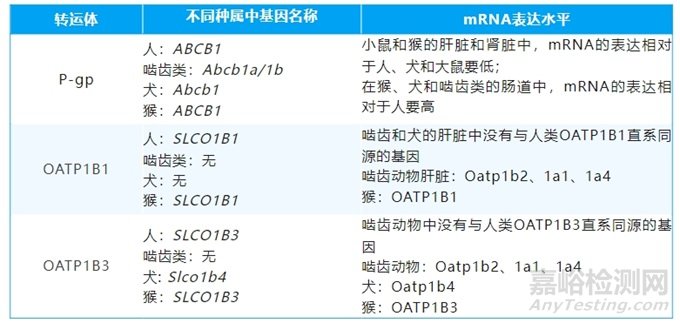
1、与P-gp抑制剂进行联合给药
P-糖蛋白(P-glycoprotein,P-gp)属于ABC转运蛋白家族,通过ATP水解获得能量把底物泵出细胞膜。该蛋白表达于多种膜屏障中,包括血脑屏障、胃肠道、肝、卵巢和胎盘等。P-gp的底物范围广泛,能够识别与转运一系列结构多样的内源性和外源性物质,包括多种化疗药物。肠道P-gp阻碍口服底物的吸收,降低其生物利用度。这种影响可通过联合使用P-gp抑制剂来降低或消除,从而促进底物的吸收,增加血药浓度。体外试验可选择的P-gp转运体抑制剂见表2。
表2. 体外试验可选择的P-gp转运体抑制剂[2]
体外测试中,常用唑喹达与Caco2或MDCK-MDR1细胞共孵育来抑制P-gp的转运作用。通过待测化合物在加与不加唑喹达的情况下外排的差异来判断其是否为P-gp的底物。临床前动物体内PK实验中,依克立达联合用药最为常见,一般提前口服灌胃,啮齿类动物给药剂量常见30-100 mg/kg,犬和猴子给药剂量约30 mg/kg。恩喹达(encequidar)也用于抑制不同的动物种属的体内P-gp活性,给药剂量与依克立达相比偏低,约10-50 mg/kg。
依克立达或恩喹达的联合给药在临床上也有应用。经典的案例是与紫杉醇类药物的联合用药来开发口服紫杉醇类药物。由于紫杉醇类药物的水溶性差且是P-gp外排的底物,口服给药严重阻碍其肠道吸收,临床上只能静脉给药。紫杉醇类药物与依克立达[3]或恩喹达[4]的联合口服可增加紫杉醇类药物的血浆浓度,使得口服紫杉醇类药物成为可能。
中枢神经系统疾病的治疗药物需要进入大脑发挥作用,但血脑屏障(blood-brain barrier,BBB)的存在限制了药物的通过,P-gp在BBB中高度表达,是许多药物不能进入大脑的原因。如何克服BBB上外排转运体如P-gp的外排,成为中枢神经系统药物治疗的瓶颈。在研发早期阶段,为了验证中枢神经系统药物的药效,通常使用P-gp基因敲除的动物开展药效实验,这为临床前药效实验的开展提高了难度和成本。而使用野生型动物与P-gp抑制剂联合用药不失为一个有效途径。拉帕替尼是一种人表皮受体2(HER2)和EGFR的小分子酪氨酸激酶抑制剂,目前已被批准用于治疗HER2阳性的晚期和转移性乳腺癌。P-gp和乳腺癌抗性蛋白(BCRP)严重影响了拉帕替尼的脑渗透作用。在大鼠中将其与依克立达联用后,依克立达显著增加了拉帕替尼在脑脊液和脑组织中的渗透(Cmax分别增加136.4%和54.7%,AUC分别增加53.7%和86.5%)[5]。
2、与OATP1B1/1B3抑制剂联合给药
有机阴离子转运多肽(organic anion transporting polypeptides,OATPs)属于溶质转运体家族,主要在肝、肾和小肠中表达,调节许多内源性和外源性化合物的组织摄取。其中OATP1B1和OATP1B3主要分布于肝细胞基底侧,在许多药物肝摄取过程中发挥重要作用,包括临床常用药物如羟甲基戊二酰辅酶A还原酶的抑制剂(他汀类)、血管紧张素Ⅱ受体拮抗剂(沙坦类)、血管紧张肽转换酶抑制剂和抗糖尿病类药物(列奈类)等均是OATP1B1和OATP1B3转运体的底物。对于主要经肝代谢的药物,肝摄取转运体介导转运活性的抑制会改变其血药浓度,继而可能增加毒副作用。十九世纪末发生西立伐他汀与吉非贝齐联用,导致多人死亡,也最终导致西立伐他汀退市[6],一些临床实验可选择的OATP1B1/1B3转运体抑制剂见表3。
体外实验中,待测药物在HEK293-MOCK细胞和OATP1B1转染的HEK293细胞中孵育时,通过加或不加环孢素A(OATP1B1抑制剂),检测待测药物摄取的差异,以评价待测药物是否是OATP1B1的底物。OATP1B3的底物评价流程与此类似。
若明确目标药物为OATP1B1/1B3底物,则可选用合适的OATP1B1/1B3抑制剂进行动物体内联用给药,考察药物在OATP1B1/1B3转运体被抑制后体内的暴露情况。由于啮齿类动物中无人的OATP1B1/1B3同源相关基因,需要使用人OATP1B1/1B3转基因的啮齿动物开展实验[7]。食蟹猴的OATP1B1/1B3氨基酸序列与人的序列相似度分别为91.9%和93.5%。体外摄取实验表明食蟹猴的OATP1B1/1B3和人的表现出相似的底物摄取速率,体外抑制试验中六种已知的人的OATP1B1/1B3抑制剂对食蟹猴的OATP1B1/1B3也表现出相似的IC50值。在体内PK实验中,利福平联合阿伐他汀(OATP1B1/1B3底物)给药后食蟹猴体内阿伐他汀暴露量改变情况与人体联合给药后情况相似。这些实验无不表明食蟹猴是临床前评价OATP1B1/1B3介导的药物相互作用的良好模型[8]。临床前体内联合给药实验设计见表4所示。
表3. 临床实验可选择的OATP1B1/1B3转运体抑制剂[2]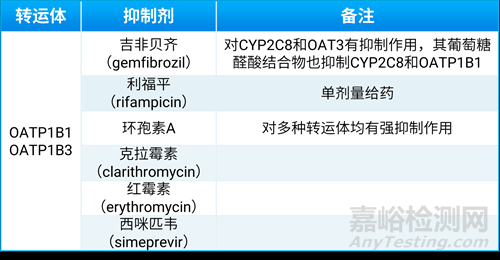
表4. OATP1B1/1B3介导的药物相互作用临床前PK实验设计[7-8]
2、与代谢酶抑制剂进行联合给药
药物吸收到体内后会被药物代谢酶代谢,发生I相代谢如氧化、还原、水解反应,以及II相代谢如葡萄糖醛酸结合、谷胱甘肽结合和氨基酸结合等。细胞色素酶P450(CYP450)是主要的药物代谢酶系,其中CYP3A4是最重要的一个亚家族,具有广泛的底物特异性,其参与了超过50%的上市小分子药物的代谢过程,占肝脏CYP酶总量的30%[9-10]。抑制相应的代谢酶会降低酶的活性,降低药物的代谢速度,从而提高血浆药物浓度,增强药效。酶诱导剂会增加酶的活性,药物的代谢速度加快,会降低血浆中的药物浓度,从而降低药效。
不同种属间CYP450酶一级结构的氨基酸序列微小差异导致了较大的底物特异性和催化活性的差别,从而造成药物代谢的差异(表5)[11]。据报道,犬的CYP2D活性与人最相似,猴的CYP2C与人最接近,小鼠的CYP1A与人较接近,小鼠和雄性大鼠的CYP3A活性与人最接近[12-13]。
表5. CYP酶家族在不同动物种属中的差异[11]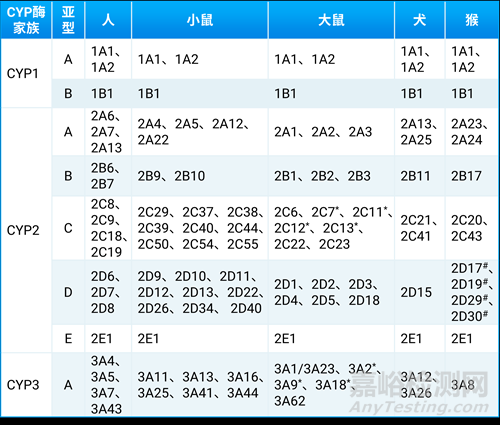
*性别差异;#品系差异
1、与CYP450酶抑制剂联合给药
氨基苯并三唑(1-aminobenzotriazole,简称1-ABT或ABT)是非特异且不可逆的CYP450酶抑制剂,因此对CYP450各个亚型的抑制时间持续较长,可以和经CYP450酶代谢的药物联用,以降低或避免CYP450的代谢。一些临床试验可选择的CYP特异性抑制剂如表6所示。
表6. 临床试验可选择的CYP特异性抑制剂[2]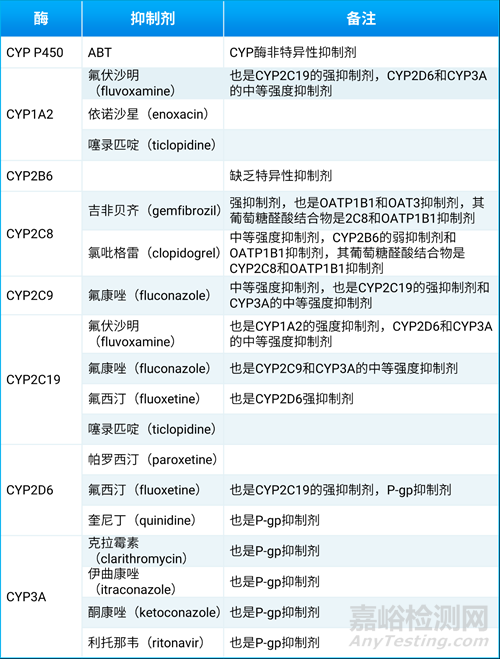
在体外实验中,可以在孵育体系中(肝微粒体、重组酶等),在加和不加ABT的情况下预先孵育30分钟,比较代谢情况,以评估ABT对CYP酶的影响[14]。动物体内实验中,给予一定剂量的ABT后,也能起到抑制CYP450酶活性的作用。ABT适应用不同的动物种属,一般提前2小时灌胃给药,啮齿类动物的给药剂量约为50-100 mg/kg。犬的给药剂量约20 mg/kg,猴的给药剂量约100 mg/kg[14]。约5-50 mg/kg的利托那韦可抑制啮齿类动物的体内CYP450酶活性。约30 mg/kg的考比司他(cobicistat)可抑制犬和猴的体内CYP450酶活性。
利托那韦和考比司他已经在临床上使用。例如,治疗新冠的复方制剂帕罗韦德(paxlovid)中,利托那韦作为奈玛特韦(nirmatrelvir)的药效增强剂,治疗HIV感染的考阿扎那韦(atazanavir)或地瑞那韦(darunavir),联用考比司他以抑制CYP3A代谢。
2、与CYP450酶诱导剂联合给药
人类的CYP1A1、CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19和CYP3A4酶是可诱导的,而CYP2D6不可被诱导[11]。酶诱导的发生是由于转录激活导致mRNA水平升高随后蛋白表达增加,核受体(PXR、CAR、GPCR等)介导CYP3A和CYP2B的转录活化,芳香烃受体(AhR)介导CYP1A的转录活化。与以上机理不一样的是CYP2E1,其通过稳定蛋白质的方式产生诱导。临床实验可选择的CYP酶诱导剂见表7。
由于酶诱导需要蛋白合成,往往需要几天时间。临床前动物PK实验中,需要连续每天给一定剂量的诱导剂,如需要连续6天给予一定剂量的利福平后才会在猴体内产生CYP酶的诱导。
另外需要注意的是,大鼠的CYP3A1(大鼠肝脏中主要的CYP3A形式)不能被人CYP3A诱导剂利福平所诱导,因此大鼠不适合于临床前CYP3A的诱导实验研究[11]。
表7. 临床试验可选择的CYP酶诱导剂[2]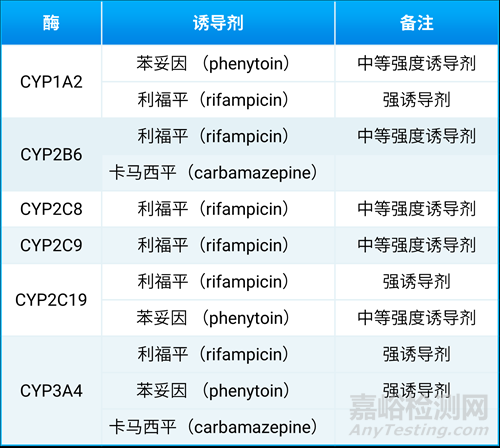
3、与酰胺水解酶抑制剂联合给药
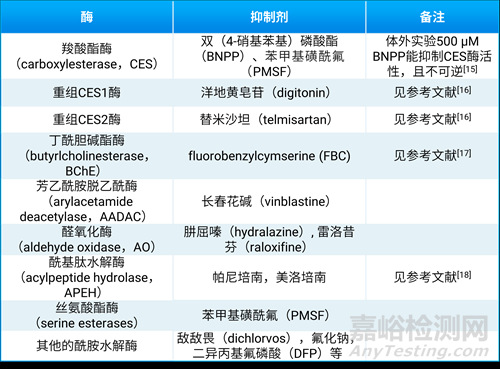
酰胺键广泛存在于药物结构中,其易受酰胺水解酶代谢而发生断裂,如图1所示。一些常见的酰胺水解酶包括羧酸酯酶(carboxylesterase,CES)、丁酰胆碱酯酶(butyrlcholinesterase,BChE)、芳乙酰胺脱乙酰酶(arylacetamide deacetylase,AADAC)、醛氧化酶(aldehyde oxidase, AO)等。其相应的常见酶抑制剂见表8。
不同动物种属在药物代谢酶的同工型组成、表达和催化活性方面的不同,对于化合物的代谢可能会造成巨大差异。人类的CES酶主要有6个亚型,其中人体中最常见的为CES1(主要分布在肝脏和肺)和CES2(主要分布在小肠和肾脏)。在人、犬和猴子的血浆中缺乏CES酶,但小鼠和大鼠血液中CES酶含量较高[19]。因此CES酶底物在小鼠和大鼠血浆中很不稳定,而在大动物或人的血浆中不存在这个问题,表现出明显的种属差异。啮齿动物的PK实验中,提前0.5小时给予一定剂量的BNPP后,由于BNPP是CES酶的不可逆抑制剂,起效后对CES酶的抑制时间较长,能明显降低CES酶的体内代谢。
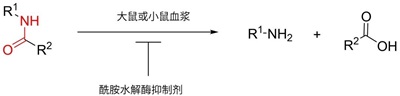
图1. 含酰胺键药物在大鼠或小鼠血浆中的稳定性
表8. 常见的酰胺水解酶抑制剂
4、与GST酶抑制剂联合给药
谷胱甘肽S-转移酶是自然界中一类最重要的解毒酶之一,其催化谷胱甘肽(GSH)和各种内源性和外源性底物的结合,以降低底物的活性和增加其溶解度。根据GST酶在细胞中的位置,GST酶可分为三大类:胞质GST酶、线粒体GST酶和微粒体GST酶。
胞质GST酶可以进一步细分为7类,包括alpha、mu(GST-M)、pi、theta(GST-T)、zeta、omega和sigma类[20],他们具有不同但重叠的底物特异性。人类和啮齿动物之间的谷胱甘肽结合存在显著种属差异和性别差异。如小鼠的总胞质GST酶、GST-M、GST-T和微粒体GST酶活性明显高于人类,大鼠的GST- M和GST-T活性明显高于人类。雄性大鼠的GST-M和GST-T活性比雌性大鼠更高。因此,如果GST酶介导的代谢是主要代谢途径,临床前研究中动物种属的选择也很关键[21]。
依他尼酸(ethacrynic acid,EA,又称“利尿酸”)是FDA批准的利尿剂(结构见图2),其也是一种有效的通过共价起效但可逆的GST酶抑制剂[22]。其他的GST酶抑制剂还包括阿魏酸松柏酯(coniferyl ferulate,CF)[23]等。
体外实验中,为了抑制GST介导的GSH结合反应,如在肝细胞或胞质中孵育时可以加入一定浓度的依他尼酸[24]。体内实验中EA联合给药的实验设计如表9所示[24]。
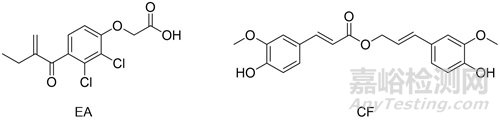
图2. 依他尼酸和阿魏酸松柏酯结构
表9. GST酶抑制剂EA联合给药临床前PK实验设计[24]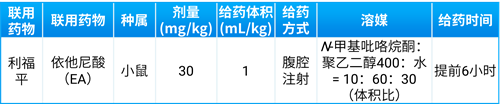
结语
总体而言,在临床前药代动力学实验中联合给药时,需要同时考虑到药物发挥作用的机制、起效部位和起效时间等,从而设计联合使用的两个药物的剂量、给药方式和给药间隔,以达到预期的实验结果。
参考文献:
[1] Chu X, Bleasby K, Evers R. Species differences in drug transporters and implications for translating preclinical findings to humans. Expert Opin Drug Metab Toxicol. 2013 Mar;9(3):237-52.
[2] 药物相互作用研究技术指导原则(试行),2021年1月。
[3] Hendrikx JJ, Lagas JS, Wagenaar E, Rosing H, Schellens JH, Beijnen JH, Schinkel AH. Oral co-administration of elacridar and ritonavir enhances plasma levels of oral paclitaxel and docetaxel without affecting relative brain accumulation. Br J Cancer. 2014 May 27;110(11):2669-76.
[4] Jackson CGCA, Hung T, Segelov E, Barlow P, Prenen H, McLaren B, Hung NA, Clarke K, Chao TY, Dai MS, Yeh HT, Cutler DL, Kramer D, He J, Zhi J, Chan WK, Kwan R, Deva S. Oral paclitaxel with encequidar compared to intravenous paclitaxel in patients with advanced cancer: A randomised crossover pharmacokinetic study. Br J Clin Pharmacol. 2021 Dec;87(12):4670-4680.
[5] Karbownik A, Sobańska K, Płotek W, Grabowski T, Klupczynska A, Plewa S, Grześkowiak E, Szałek E. The influence of the coadministration of the p-glycoprotein modulator elacridar on the pharmacokinetics of lapatinib and its distribution in the brain and cerebrospinal fluid. Invest New Drugs. 2020 Jun;38(3):574-583.
[6] Maeda K. Organic anion transporting polypeptide (OATP)1B1 and OATP1B3 as important regulators of the pharmacokinetics of substrate drugs. Biol Pharm Bull. 2015;38(2):155-68.
[7] LeBel M, Masson E, Guilbert E, Colborn D, Paquet F, Allard S, Vallée F, Narang PK. Effects of rifabutin and rifampicin on the pharmacokinetics of ethinylestradiol and norethindrone. J Clin Pharmacol. 1998 Nov;38(11):1042-50.
[8] Shen H, Yang Z, Mintier G, Han YH, Chen C, Balimane P, Jemal M, Zhao W, Zhang R, Kallipatti S, Selvam S, Sukrutharaj S, Krishnamurthy P, Marathe P, Rodrigues AD. Cynomolgus monkey as a potential model to assess drug interactions involving hepatic organic anion transporting polypeptides: in vitro, in vivo, and in vitro-to-in vivo extrapolation. J Pharmacol Exp Ther. 2013 Mar;344(3):673-85.
[9] Ince I, Knibbe CA, Danhof M, et al. Developmental changes in the expression and function of cytochrome P450 3A isoforms: evidence from in vitro and in vivo investigations [J]. Clin Phar‐
macokinet, 2013, 52: 333-345.
[10] Zhou SF. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4 [J]. Curr Drug Metab, 2008, 9: 310-322.
[11] Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2006 Dec;2(6):875-94.
[12] BOGAARDS JJ, BERTRAND M, JACKSON P et al.: Determining the best animal model for human cytochrome P450 activities: a comparison of mouse, rat, rabbit, dog, micropig, monkey and man.
Xenobiotica (2000) 30:1131-1152
[13] ZUBER R, ANZENBACHEROVA E, ANZENBACHER P: Cytochromes P450 and experimental models of drug metabolism. J. Cell Mol. Med. (2002) 6:189-198.
[14] (a) Balani SK, Zhu T, Yang TJ, Liu Z, He B, Lee FW. Effective dosing regimen of 1-aminobenzotriazole for inhibition of antipyrine clearance in rats, dogs, and monkeys. Drug Metab Dispos. 2002 Oct;30(10):1059-62. (b) Balani SK, Li P, Nguyen J, Cardoza K, Zeng H, Mu DX, Wu JT, Gan LS, Lee FW. Effective dosing regimen of 1-aminobenzotriazole for inhibition of antipyrine clearance in guinea pigs and mice using serial sampling. Drug Metab Dispos. 2004 Oct;32(10):1092-5.
[15] Eng H, Niosi M, McDonald TS, Wolford A, Chen Y, Simila ST, Bauman JN, Warmus J, Kalgutkar AS. Utility of the carboxylesterase inhibitor bis-para-nitrophenylphosphate (BNPP) in the plasma unbound fraction determination for a hydrolytically unstable amide derivative and agonist of the TGR5 receptor. Xenobiotica. 2010 Jun;40(6):369-80.
[16] Shimizu M, Fukami T, Nakajima M, Yokoi T. Screening of specific inhibitors for human carboxylesterases or arylacetamide deacetylase. Drug Metab Dispos. 2014 Jul;42(7):1103-9.
[17] Kamal MA, Shakil S, Nawaz MS, Yu QS, Tweedie D, Tan Y, Qu X, Greig NH. Inhibition of Butyrylcholinesterase with Fluorobenzylcymserine, An Experimental Alzheimer's Drug Candidate: Validation of Enzoinformatics Results by Classical and Innovative Enzyme Kinetic Analyses. CNS Neurol Disord Drug Targets. 2017;16(7):820-827.
[18] Suzuki E, Nakai D, Yamamura N, Kobayashi N, Okazaki O, Izumi T. Inhibition mechanism of carbapenem antibiotics on acylpeptide hydrolase, a key enzyme in the interaction with valproic acid. Xenobiotica. 2011 Nov;41(11):958-63.
[19] Di L. The Impact of Carboxylesterases in Drug Metabolism and Pharmacokinetics. Curr Drug Metab. 2019;20(2):91–102.
[20] Mannervik B, Board PG, Hayes JD, Listowsky I, Pearson WR. Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 2005;401:1-8.
[21] Doerksen MJ, Seo D, Smith AD, Jones RS, Coughtrie MWH, Collier AC. Comparisons between human and rodent hepatic glutathione S-Transferase activities reveal sex and species differences. Xenobiotica. 2023 Apr;53(4):223-230.
[22] Ploemen, J. H.; van Ommen, B.; Bogaards, J. J.; van Bladeren, P. J., Ethacrynic Acid and
its Glutathione Conjugate as Inhibitors of Glutathione S-transferases. Xenobiotica 1993, 23
(8), 913-923.
[23] Chen C, Wu C, Lu X, Yan Z, Gao J, Zhao H, Li S. Coniferyl Ferulate, a Strong Inhibitor of Glutathione S-Transferase Isolated from Radix Angelicae sinensis, Reverses Multidrug Resistance and Downregulates P-Glycoprotein. Evid Based Complement Alternat Med. 2013;2013:639083.
[24] Yang X, Ong HW, Dickmander RJ, Smith JL, Brown JW, Tao W, Chang E, Moorman NJ, Axtman AD, Willson TM. Optimization of 3-Cyano-7-cyclopropylamino-pyrazolo[1,5-a]pyrimidines toward the Development of an In Vivo Chemical Probe for CSNK2A. ACS Omega. 2023 Oct 10;8(42):39546-39561.

来源:药明康德DMPK


