我们都知道,FDA有权对生产FDA监管产品的制药公司进行突击或预知检查,并将其意见记录在FDA 483表格上,通常缩写为“483”。483 中引用的法规领域的观察总结,可以在 FDA 主页的“检查观察Inspection Observations”小节中的电子表格获得。除了观察内容之外,被引用的次数是这个表格透露的最直接的信息。
药物稳定性研究是一门成熟的学科,当然也是一项重要的监管要求。然而在 2019 年至 2021 财年的许多 FDA 483 中都提到了稳定性计划的缺陷。FDA 现已公布了 2022 财年(2021年10月至 2022年9月)的数据。在“药物”领域发布了 466 个 FDA 483。评估表明,稳定性计划中的缺陷再次成为重灾区。
在“药物”领域,FDA过去几年发布的报告数量如下:
2019 财年:779 份 FDA 483 份表格
2020 财年:349 份 FDA 483 份表格
2021 财年:215 份 FDA 483 份表格
2022 财年:466 份 FDA 483 份表格
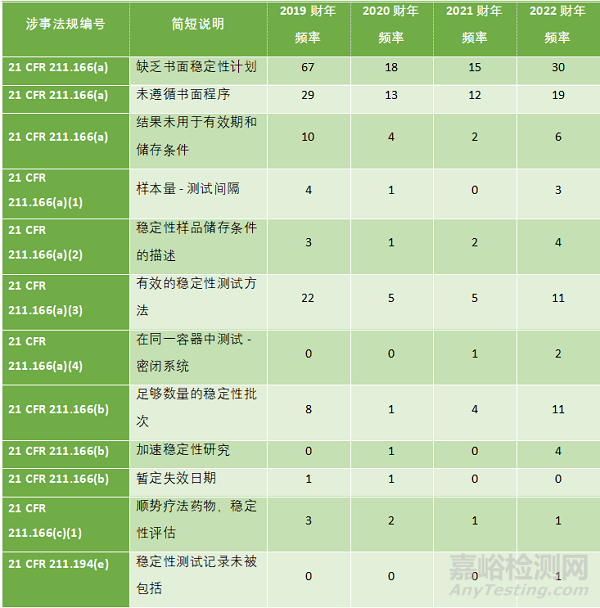
表1概述了与稳定性测试相关的典型缺陷。[1] 可以看出,在上一财年的检查中,几乎对 21 CFR Part 211.166 的所有子段落都有涉事项目。
表1. FDA2022财年483表涉及稳定性测试的观察总结
二、药物稳定性研究的监管要求
稳定性研究的要求在联邦法规(Code of Federal Regulations)中有明确定义,主要体现在 21 CFR 211.166 条中:
1、211.166 稳定性测试
(a) 应准备药品稳定性特征的书面测试程序。这种稳定性测试的结果应用于确定适当的储存条件和有效期。应遵循的书面计划包括:
基于统计标准的样本大小和测试间隔,用于检查的每个属性,以确保对稳定性的有效估计;
留样保存条件;
可靠、有意义、具体的测试方法;
在与上市药品相同的容器密闭系统中对成品药进行测试;
在规定配药时间(按照标签中的指示)检测成品药的重构(reconstitution),以及在重构后进行检测。
(b) 应对每种药品进行足够数量的批次测试,以确定适当的有效期,并保存此类数据的记录。结合相关成分、药品和容器密闭系统所进行的加速研究,可用于支持暂定有效期,但前提是完整的保质期研究结果暂不可完成,但正在进行中。如果使用加速研究的数据来预测暂定有效期,而预测的暂定有效期超出数据支持的实际保质期,则必须进行稳定性研究,包括在适当的时间间隔进行药品测试,直到暂定有效期得到验证或确定适合的有效期。
(c) 对于顺势疗法药品的要求如下:
应至少基于对药品成分相容性的测试或检查,并根据药品的销售经验,表明该药品在正常情况下没有降解,对稳定性进行书面评估或预估使用期限。
稳定性评价应基于与上市药品相同的容器密闭系统。
(d) 标有“无美国效力标准No U.S. Standard of Potency”的过敏性提取物不受21 CFR 211.166要求的约束。
另外,21 CFR 211.194中也提到了稳定性测试:
2、211.194 实验室记录
(e)应保存根据 § 211.166 执行的所有稳定性测试的完整记录。
三、药物稳定性测试要素
决定如何以及何时对新药进行稳定性测试,是药物研发和生命周期管理中的关键内容之一。稳定性测试提供了原料药或成品药的质量与时间和保存环境之间的关联。它还规定了原料药的复验期、成品药的保质期,以及推荐的储存条件。[3] FDA 的稳定性测试要求与欧盟和日本协调并保持高度一致,因此在欧盟、日本或美国这三个地区中的任何一个地区生成的稳定性信息将被其他两个地区相互接受。
1、原料药稳定性试验的关键要素如下
(a) 压力测试(stress testing):原料药的压力测试可以帮助识别可能的降解产物,这反过来可以帮助建立降解途径和分子的内在稳定性,并验证所用分析程序的稳定性指示能力。压力测试的性质将取决于原料药和涉及的成品药类型。
压力测试可能对单批原料药进行。它应包括温度(以 10°C 的增量(例如,50°C、60°C 等)高于加速测试的温度)、适当的湿度(例如,75% RH 或更高)、氧化和对原料药进行光解。测试还应评估药物物质在溶液或悬浮液中,在广泛的 pH 值范围内对水解的敏感性。光稳定性测试应该是压力测试的一个组成部分。ICH Q1B 中描述了光稳定性测试的标准条件。
在压力条件下检查降解产物,有助于建立降解途径,以及开发和验证合适的分析程序。但是,如果已经证明某些降解产物不是在加速或长期储存条件下形成的,则可能没有必要专门检查它们。
这些研究的结果将构成提供给监管机构的信息的组成部分。
(b) 批次的选择:至少应使用三个primary批次(primary批次是指用于稳定性研究的一批活性药物成分或成品药物,稳定性数据在注册申请中提交,用于建立复验期或保质期;primary批次的制造规模至少需要与中试(pilot scale)中的最小规模相当)的原料药进行正式的稳定性试验。其生产过程必须与用于生产批次的最终过程相同。进行正式稳定性研究的原料药批次的总体质量应能代表生产规模生产的原料药的质量。
(c) 容器密闭系统:必须对包装在与最终包装相同的容器密闭系统中的药物进行测试。
(d) 测试频率:测试频率应保证建立药物稳定性特征。对于建议复验期至少为 12 个月的原料药,在长期贮存条件下的试验频率通常应为第一年每 3 个月一次,第二年每 6 个月一次,此后每年一次直至建议的复验期。
在加速储存条件下,在6 个月的研究框架下,建议至少三个时间点,包括初始和最终时间点(例如,0、3 和 6 个月)。如果存在加速研究的结果可能接近显著变化标准的预期(基于开发经验),则应通过在最后时间点添加样本,或通过在研究设计中包括第四个时间点来进行增加的测试。
(e) 当由于加速储存条件发生显著变化而要求在中期储存条件下进行测试时,至少应该包括四个时间点,包括初始和最终时间点(例如,0、6、9、12 个月),从建议时间开始进行为期 12 个月的研究。
(f) 存储条件:测试必须评估存储条件,以测试其热稳定性和对水分的敏感性。这些条件还必须涵盖储存、运输和后续使用。
(g) 规格Specification:ICH Q6A 和 Q6B 中阐述了规格,即测试内容、分析方法和建议的验收标准。此外,Q3A 中讨论了原料药中降解产物的规格。稳定性研究应包括对原料药在储存过程中容易发生变化,并对那些可能影响质量、安全性和疗效的那些属性进行测试。测试应酌情涵盖物理、化学、生物学和微生物学属性。应采用经过验证的稳定性分析程序。是否应该进行重复分析以及重复分析的程度取决于验证研究的结果。
2、成品药稳定性试验的关键要素如下
(a) 成品药正式稳定性研究应基于对原料药行为和特性的了解,以及原料药的稳定性研究和临床制剂研究中获得的经验。应说明在正式稳定性研究中可能发生的储存变化和选择测试属性的理由。
(b) 光稳定性测试:如果合适,应至少对一个初级批次的药品进行光稳定性测试。ICH Q1B 中描述了光稳定性测试的标准条件。
(c) 批次选择:应提供至少三个primary批次药品 (primary批次是指用于稳定性研究的一批活性药物成分或成品药物,稳定性数据在注册申请中提交,用于建立复验期或保质期) 的稳定性研究数据。primary批次应具有相同的配方,并包装在与上市销售相同的容器密闭系统中。用于primary批次的制造过程应模拟生产批次的工艺,并应提供与销售产品具有相同质量和符合相同规格的产品。三批中的两批至少应是中试规模批次,如果有恰当理由,第三批可以小一些。在可能的情况下,药品的批次应使用不同批次的原料药生产。稳定性研究应对药品的每个单独规格和容器尺寸进行,除非使用了涵括设计(bracketing)或矩阵设计(matrixing)。
(d) 容器封闭系统:稳定性测试应在拟上市销售的容器密闭系统中进行(包括任何二级包装和容器标签)。
(e) 规格Specification:ICH Q6A 和 Q6B 中阐述了specification,即测试列表、分析程序参考和建议的验收标准,包括放行和保质期规范的不同验收标准的概念。此外,药品中降解产物的规格在 Q3B 中进行了说明。
稳定性研究应包括测试药品在储存过程中易发生变化并可能影响质量、安全性和疗效的那些属性。测试应酌情包括物理、化学、生物和微生物属性、防腐剂含量(例如抗氧化剂、抗菌防腐剂)和功能测试(例如剂量输送系统)。分析程序应经过充分验证。是否应该进行重复分析以及重复分析的程度取决于验证研究的结果。
(f) 测试频率:对于长期研究,测试频率应足以确定药品的稳定性。对于建议保质期至少为 12 个月的产品,长期储存条件下的测试频率通常应为第一年每 3 个月一次,第二年每 6 个月一次,此后在建议保质期内每年一次。
在加速储存条件下,建议至少进行 6 个月的研究,并至少涵盖三个时间点,包括初始和最终时间点(例如,0、3 和 6 个月)。如果预期(基于开发经验)加速测试的结果可能接近显著变化标准,则应通过在最后时间点添加样本,或通过在研究设计中包括第四个时间点来进行增加测试。
四、药物稳定性研究条件
1、原料药
(a) 常规条件存储的原料药
*由申请人决定是否在 25 ± 2°C/60% 相对湿度± 5% 相对湿度,还是30°C ± 2°C/65% 相对湿度± 5%相对湿度下进行长期稳定性研究。
*如果 30°C ± 2°C/65% 相对湿度± 5%相对湿度被选作长期条件,则没有中期研究。
如果选择25°C ± 2°C/60% 相对湿度 ± 5% 相对湿度条件的长期研究,并且在加速存储条件下 6 个月测试期间的任何时间出现了“显著变化”,则应在中期存储条件下进行额外测试,并且应根据重大变更标准进行和评估。中期储存条件下的测试应包括所有测试,除非另有证明。初始申请应包括至少 6 个月的数据,这些数据来自在中间储存条件下进行的为期 12 个月的研究。原料药的“显著变化”被定义为不符合其质量标准(OOS)。
(b) 冰箱refrigerator内存储的原料药
如果在加速贮存条件下 3 到 6 个月的测试之间发生显著变化,建议的复试期应基于长期贮存条件下可用的实时数据。如果在加速储存条件下的前 3 个月测试中发生显著变化,则应提供讨论,以解决标签储存条件之外的短期偏移的影响,例如在运输或处理过程中。如果合适,可以通过对单个批次的原料药进行短于 3 个月、但比平时更频繁的测试,进一步测试来支持这种讨论。如果在前 3 个月内发生了显著变化,则没有必要继续测试原料药 6 个月。
(c) 拟冷藏在freezer中的原料药
对于拟在freezer中贮存的原料药,复验期应基于在长期贮存条件下获得的实时数据。对于拟储存freezer中的原料药,在没有加速储存条件的情况下,应在升高的温度(例如 5°C ± 3°C 或 25°C ± 2°C)环境下,对单个批次进行适当的测试,以确定短期偏离建议标签存储条件的时间段,例如在运输或处理过程中。
(d) 拟在 -20°C 以下储存的原料药
拟在 -20°C 以下储存的原料应根据具体情况进行处理。
2、成品药
(a) 常规条件存储的成品药
*由申请人决定是否在 25 ± 2°C/60% 相对湿度± 5% 相对湿度,还是30°C ± 2°C/65% 相对湿度± 5%相对湿度下进行长期稳定性研究。
*如果 30°C ± 2°C/65% 相对湿度± 5%相对湿度被选作长期条件,则没有中间条件。
如果在 25°C ± 2°C/60% RH ± 5% RH 下进行长期研究,并且在加速存储条件下 6 个月测试期间的任何时间出现“显著变化”,中期存储条件下进行的额外测试应根据重大变更标准进行和评估。初始申请应包括至少 6 个月的数据,这些数据来自在中期储存条件下进行的为期 12 个月的研究。
一般来说,药品的“显著变化”定义为:
测定值较初始值有 5% 的变化;或在使用生物或免疫程序时未能达到效力的验收标准;
降解产物超标;
未能满足外观、物理属性和功能测试的验收标准(例如,颜色、相分离、再悬浮性、结块、硬度、每次启动的剂量输送);然而,在加速条件下可能会发生一些物理属性的变化(例如,栓剂软化,乳膏融化);
pH值不符合验收标准;
12个剂量单位溶出度不合格。
(b) 拟储存在冰箱refrigerator中的成品药
如果药品包装在半透性容器中,应提供适当的信息以评估失水程度。
如果在加速储存条件下 3 至 6 个月的测试中发生显著变化,则建议的保质期应基于长期储存条件下可用的实时数据。如果在加速储存条件下的前 3 个月测试中发生显著变化,则应提供讨论,以解决标签储存条件之外的短期偏移的影响,例如在运输和处理过程中。如果合适,可以通过在短于 3 个月、但比平时更频繁的测试期间,对单批药品进行进一步测试来支持这种讨论。如果在前 3 个月内发生了显著变化,则没有必要继续测试产品 至6 个月。
(c) 拟冷藏于freezer的成品药
对于拟在freezer中储存的药品,保质期应基于在长期储存条件下获得的实时数据。对于拟储存在freezer中的药品,在没有加速储存条件的情况下,应在高温(例如 5°C ± 3°C 或 25°C ± 2°C)下对单个批次进行适当的测试,并确定一个时间段,以解决在建议的标签存储条件之外的短期偏移的影响。
(d) 拟在 -20°C 以下储存的成品药
拟在 -20°C 以下储存的药品应根据具体情况进行处理。
参考文献:
[1] ECA Academy. In 2022 again Numerous FDA 483s due to Deficiencies in the Stability Program. 07. 12. 2022
[2] ICH. ICH Harmonised Tripartite Guideline Stability Testing of New Drug Substances and Products Q1A(R2). 06. 02. 2003.
[3] FDA (2003) Guidance for Industry Q1A(R2) Stability Testing of New Drug Substances and Products retrieved on 05/06/2021 from https://www.fda.gov/media/71707/download