您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-07-06 22:36
1. 介绍
杂质根据其来源可分为加工、降解或外来杂质(污染或外来杂 质)。外来杂质是有机或无机的化学实体,在加工、储存或交付 过程中被引入原料药或制剂中[1]。 与合成的、降解的或赋形剂相 关的杂质相比,外来杂质可以代表多种化学结构和化合物类别, 并且可以以广泛变化的浓度存在[2–5]。在更广泛的外来杂质类别 中,有一部分杂质会在接触容器密闭或包装系统中使用的材料、 临床使用或预装注射器中的储存材料时迁移到药品中[1,6]。这些 可萃取物/可浸出物可能会影响产品质量和/或安全性,从而对患者造成风险[7,8]。这些可萃取物/浸出物的表征和控制仍然是产品开发 团队面临的重大挑战 [1,9]。要记录所有可提取物和可浸出杂质的报告,可提取物和可浸出物安全信息交换 (ELSIE) 成立于 2007 年,该数据库包含迄今为止报告的 477 种此 类可提取物/可浸出物的信息[10]. 国际人用药物技术要求协调委员 会 (ICH) 还发布了题为“杂质:药物和生物制品可提取物和可浸出物 的评估和控制”的 Q3E 指南[11] 用于跟踪此类杂质。
有几项研究详细说明了从容器密闭/输送系统、医疗器械和其他组件(例如用于加工、储存和/或输送药物的预装注射器、垫圈、塞子 和塑料储存袋)中提取和可浸出杂质的结果 [1–5,12–14]. 最近,预充式注射器作为药物输送系统获得了广泛的认可,据报道,这些预充式注射器中的许多潜在可萃取物/可浸出物[1,12,13]。一次性塑 料注射器正被广泛用于药物分析实验室,目前的研究是为了鉴定来自内部发现化合物的药物产品方法开发过程中用于样品过滤的注射器的杂质。据观察,相对保留时间 (RRT) 0.96 处的杂质峰面积百分比是与 100 mg 片剂相比,12.5 mg 强度片剂的含量显着更高(表 S1)。
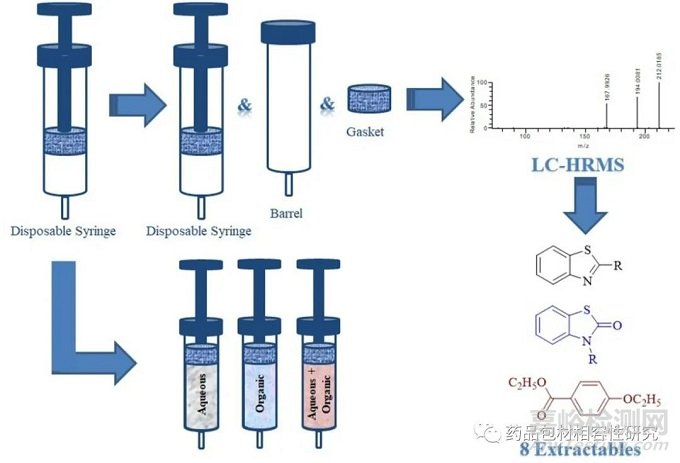
这两种制剂的样品制备(提取、超声和过滤后)的主要区别在于稀释,即 100 mg 规格的片剂包括 10 倍稀释,而 12.5 mg 规格的片剂溶液直接注入高效液相色谱 (HPLC)无需稀释。因此, 进行了详细调查以确定该杂质的来源。在稀释剂中制备的样品、 安慰剂通过 0.45 µm 聚四氟乙烯 (PTFE) 滤膜过滤、离心安慰剂和从注射器冲洗的稀释剂(图 S1)证实一次性注射器是外来杂质的来源。在有关一次性注射器可提取物/可浸出物的已发表报告中 [6,15–17], 艾奥多等人[16] 和 Salmona 等人 [17] 报道了使用蒸馏水作为提取溶剂的一次性注射器中苯并噻唑基可提取物/可浸出物的结构。据作者所知,目前缺乏关于在水和有机溶剂混合物存在下从一次性注射器中提取的可提取物的报告,这些溶剂通常在药物开发过程中的原料药和药品分析过程中用作样品稀释剂。在当前的研究中使用不同的临床级一次性注射器对此进行了探索。进行注射器组件提取以了解注射器不同部分的贡献。在此,我们报告了使用液相色谱-高分辨质谱 (LC-HRMS) , 核磁共振 (NMR) 和/或 红外 (IR) 光谱鉴定的塑料一次性注射器中的八种可提取物[18]。在八个提议的结构中,四个以前没有被报告为可提 取/可浸出,三个被发现是新的化学结构。
2. 实验设计
2.1. 化学品和试剂
不同品牌的一次性注射器(品牌 A-D)购自当地药店(印度班加罗尔)。甲醇 (MeOH)、乙腈 (MeCN)、盐酸 (HCl) 和氢氧化钠 (NaOH) 购自 Merck KGaA(德国达姆施塔特)。环氧乙烷 (EtO)、 二氯甲烷 (DCM)、 氘代氯仿 (CDCl3) 和甲酸铵购自 Sigma- Aldrich(美国密苏里州圣路易斯)。甲酸购自 Biosolve Chimie (Dieuze, France)。纯化的 Milli-Q 水(MQW,本文中称为水)由 Millipore(Bradford,PA,美国)的 Milli-Q plus 纯化系统获 得,注射用水(WFI,Baxter,Gurgaon,India)购自当地药房。
2.2. 设备
液相色谱在 Waters Alliance®-HPLC 光电二极管阵列系统 (Waters Corporation, Milford, MA, USA) 上进行。Empower 3 软 件用于数据采集和处理。X-Bridge Shield®RP18 柱,150 mm ×4.6 mm,3.5 µm (Waters Corporation) 用于样品的色谱分析。
高分辨率质谱 (HRMS) 分析在 LTQ Orbitrap Velos 质谱仪上进行,该质谱仪与 Accela Ultra HPLC 系统(均来自美国加 利福尼亚州圣何塞 Thermo Scientific)和 Agilent 1200 使用布鲁克提供的TOPSPIN 3.2 软件。IR 分析是在 PerkinElmer (Shelton, CT, USA) 的 Spectrum 100 FT-IR 仪器上进行的。
2.3. 样品制备和 HPLC 分析
对于注射器可萃取研究,(1) 注射器用选定的溶剂填充大约一 半,并在垫圈和针筒就位的情况下摇动大约 20 秒,(2) 然后将注 射器的内容物连续转移到另外四个注射器中,然后摇动约每个注 射器 20 秒,最终内容物被注入 HPLC。在针筒可提取研究的情况 下,采用了类似的程序,注射器中没有垫圈和柱塞。使用乙腈和 水的 1:1 v/v 混合物作为溶剂对品牌 A、B、D(5 mL)和品牌 C(2 mL,因为当地市场上没有该品牌的 5 mL 注射器)进行评估. 还使用相同的乙腈和水混合物 (1:1 v/v) 评估了 A 品牌的 1、2、 5 和 10 mL 容量注射器。使用注射用水、水、甲醇、 乙腈和等体 积的水与乙腈和甲醇的混合物,采用与上述类似的程序。为了从 垫圈中富集可提取物,将 A 品牌注射器的 5 mL 容量注射器的 5 个垫圈在 10 mL 乙腈和水 (1:1 v/v) 混合物中超声处理 2 分钟, 将液体内容物离心 5以每分钟 4000 转 (rpm) 的转速旋转, 上清液用于 HPLC 分析。HPLC 流动相由 10 mM 甲酸铵和 0.05 % v/v 甲酸水溶液组成(A) 和甲醇 (B) 在梯度模式下 (T min/%A: T0/90; T3/90; T18/10; T28/10; T31/90; T35/90)。在分析过程中,柱温箱温度保持在 40◦C。检测波长,流速和进样体积分别为 254 nm、1 mL/min 和 10 µL。
2.4. LC-HRMS 研究
部分列出的 HPLC 方法2.3 也用于 LC-HRMS 研究。HRMS 仪器 使用正离子模式的电喷雾电离源进行操作,源电流为 100.0 µA, 毛细管温度为 250◦C、300◦C 和 350◦ C 代表品牌 A 可提取物全扫描,品牌 B 可提取物全扫描和品牌 A 可提取物 MS/MS,分别。氮气和氦气分别用作鞘气和辅助气。MS/MS 数据是使用更高能量的 C-trap 解离 (HCD) 在数据 依赖模式下采集的,其中所需的最小信号、隔离宽度、归一化碰 撞能量、默认电荷状态和激活时间设置为 500.0、2.00、35.0 , 分别为 1 和 0.100。
2.5. 用于 NMR 和 IR 研究的可提取 E3 的富集和分离
为了获得足够的 NMR 研究样品,将大约 100 个 2 mL 品牌 A 注射器的垫圈在 100 mL 1 N NaOH 溶液中超声处理 10 分钟,并 在 60◦C 下储存 2 小时。所得提取物用 5 N HCl 中和,并用 0.45µm 聚偏二氟乙烯 (PVDF) 过滤器过滤。然后用50mL DCM萃取滤液。在旋转蒸发器上干燥DCM层。然后将干燥的残留物溶解在 0.6 mL相色谱系统( Agilent Technologies, Waldbronn, Germany) 连CDCl3中以收集1H、13C、通过偏振转移-135 (DEPT-135)、异核单接到 LTQ OrbitrapTM值质谱仪 (Thermo Scientific)。Thermo Xcalibur 软件 2.1、2.2 和 3.0 用于数据采集和处理。
在 Bruker 400 MHz(Bruker BioSpin Corporation,Fällanden, Switzerland)仪器上进行一维(一维)和二维(二维)核磁共振 实验。所有数据都已获取使用与上面列出的类似程序进行提取以获得更高的样品量。使用 衰减全反射 (ATR) 采样技术从 4000 cm−1到 400 cm−1扫描样品。
3. 结果和讨论
进行了详细调查,以确定来自不同品牌一次性注射器的潜在可浸出物(可提取物)。为此,对四个品牌 (A-D) 的一次性注射器 进行了评估。其中,显示出大量可萃取物(A 和 B)的品牌使用 不同的溶剂进行评估,即水性、有机及其混合物。还评估了不同容量注射器的可提取物最大量的品牌 A,以了解提取倾向。最后, 对品牌 A 和 B 进行评估,以找出注射器不同部位的可提取物的确 切来源,并使用主要可提取物来源的富集样品进行识别。下面详 细讨论同样的问题。
3.1. 四种不同品牌一次性注射器的 HPLC 分析
比较了 A、B、C 和 D 品牌注射器的色谱数据在乙腈:水 (1:1 v/v) 中的可提取物,这是 HPLC 方法中最常见的稀释剂之一,在 分析方法开发和药品的验证。代表性色谱图在图 S2a 中给出。对 四个品牌的总可萃取物的面积计数(表 S2)进行了比较,并在图 1a。不同注射器品牌的提取倾向各不相同,品牌 A 的可提取物含 量最高,其次是品牌 B,即品牌 A 的约 42%。两个品牌的可提取 物特征不同,品牌 B 中不存在品牌 A 的主要可提取物反之亦然(图 S2a),这表明不同品牌的一次性注射器可能会产生不同类 型的可提取物。从品牌 B 和 C 中提取的主要成分相似,但是,在 品牌 C 的情况下,其强度明显较低(图 S2a)。在品牌 D 注射器 中未观察到可提取物,因此品牌 C(与品牌 B 相似)和品牌 D 未 进一步评估。我们还想在此提及,各种可萃取物的响应因子可能 不同。这里的目的只是为了证明不同品牌的一次性注射器对浸出 的敏感性,考虑到不同可提取物的几乎相似的响应因子。
3.2. A牌不同容量一次性注射器的可提取物倾向
由于在品牌 A 注射器中观察到最大可提取物,因此还使用等体积 的乙腈和水混合物进一步评估了注射器体积(1、2、5 和 10 mL)的 影响(图 S2b)。在所有体积中都观察到了类似的可提取曲线,而 5 和 10 mL 注射器的总面积计数相对较低(图。1b)。该结果表明,A 牌不同容量一次性注射器的各个部件的原材料很可能来自共同的来源。
3.3. 不同溶剂对可萃取物的影响
使用不同溶剂对品牌 A 和 B (5 mL) 注射器评估不同可萃取物 的萃取倾向,因为两者在初始研究中都显示出显着的可萃取物
(部分3.1). 注射用水、水、甲醇、乙腈和等体积水与乙腈和甲醇 的混合物用作溶剂,所得色谱图分别显示在图 S2c 和 S2d 中,分 别用于品牌 A 和 B。最高含量在纯有机溶剂(即乙腈和甲醇)存在下观察到可提取物的数量, 然后是它们与水的混合物(乙腈:水和甲醇:水,均为 1:1 v/v)。仅使用水性溶剂(注射用水和水)观察到极少量的可萃取物(图。1C)。在品牌 A 的情况下,当溶剂从水溶剂和有机溶剂 的混合物变为 100% 有机溶剂(有机溶剂比混合物更高)时,只有提取程度(每种可提取物的数量)发生变化,而在在品牌 B 的情况下,当使用纯乙腈和甲醇时,观察到两种新的可萃取物(图 S2d,26-27 分钟 RT)。结果强调,使用不同的溶剂和/或溶剂极性的变化可能会导致在可提取物的数量和类型方面的不同提取倾向。因此,使用一种溶剂的可萃取性研究可能无法预测使用另一种溶剂的结果,需要使用包括任何酸性或碱性改性剂在内的精确溶剂组合进行。
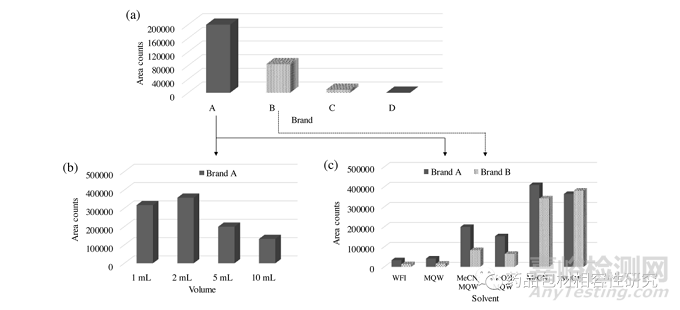
图1.不同品牌一次性注射器(a)、品牌A不同容量注射器(b)和品牌A和B的5mL注射器使用各种溶剂(c)的总可萃取物的比较面积计数柱状图。注意:提供了总面积计数作为一种手段,以证明哪个品牌/溶剂导致最大量的可提取物,假设它们的响应因子几乎相等。
关键词:MeCN,乙腈;甲醇,甲醇; MQW,Milli-Q 水和WFI,注射用水。
3.4. 评估注射器组件以确认可提取物的确切来源
在评估的一次性注射器品牌(品牌 A–D)中,品牌 A 和 B 注射 器显示出两种不同的可提取特征(部分3.1). 因此,对这两个品牌的 注射器进行了进一步审查,以破译可提取物的确切来源。为此, 使用水和乙腈 (1:1 v/v) 的混合物作为溶剂分别提取了桶、垫圈和整个注射器,所得色谱图显示在图2a和 2b。未评估柱塞,因为 它没有与溶剂直接接触。来自注射器、针筒和垫圈的比较数据表明,从注射器冲洗液中观察到的可萃取物都存在于垫圈中。桶冲洗显示可提取物的存在可忽略不计,这些可提取物在移除垫圈期间可能已从垫圈转移到桶。尽管在品牌 B 中,在桶冲洗中出现了一个额外的小峰,这不是垫圈可萃取物的一部分。除此之外,几 乎所有的可萃取物都是垫片提取物的一部分,因此使用富集的垫片提取物来鉴定主要杂质。
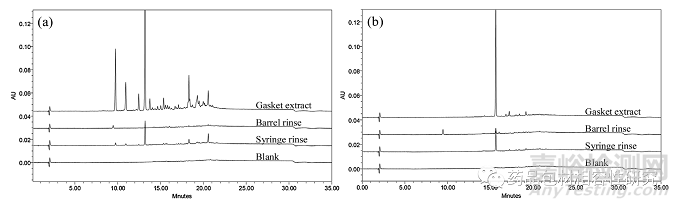
图2.品牌A(a)和品牌B(b)注射器(5mL)不同部分的比较色谱图显示出可提取物的趋势。
3.5. 可萃取物的表征
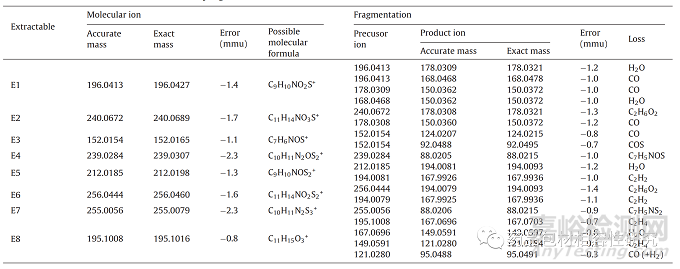
七种可萃取物,A 品牌注射器垫圈萃取物的 E1-E7 在 RRT 0.74、 0.81、0.83、0.94、1.00(RT 13.19 min 处的主峰被视为 RRT 的参考)、1.04 和 1.04(图 3a) 借助质量和/或 NMR 数据以及B 品牌注射器垫圈的主要可提取物之一进行表征(E8,RT 15.65 分钟,图 3b)。ESI + ve 模式下所有可萃取物的 HRMS 线谱如图 所示图 4和数据被编译在表格1。E1-E3、E5 和 E6 是 35 归一化 碰撞能量下的 MS/MS 谱图,而在 E4、E7 和 E8 碰撞诱导解离导 致分子离子完全消失的情况下,显示了全扫描的谱图。E4、E7 和 E8 的这些全扫描谱图显示存在与 MS/MS 谱图相同的碎片。分子离子和碎片的最佳可能分子式和精确质量是使用元素组成计算器计 算的,并包含在表格1 以及毫质量单位 (mmu) 中的误差。同表中 的数据用于建立这些可萃取物的碎裂途径。通过比较理论和观察 到的相对同位素丰度值,也证实了通过精确质量提出的化学式(图 S3 和表 2)。观察到的所有可提取物的 M + 1 和 M + 2 相 对同位素丰度值均在理论相对丰度值的 ±5% 以内。
表2. 来自品牌 A 和 B 注射器的可提取物的相对同位素丰度数据
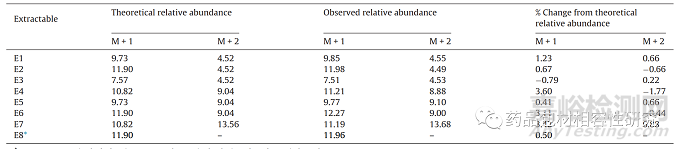
图片*M+ 2 未包括在内,因为其理论相对丰度百分比小于 1
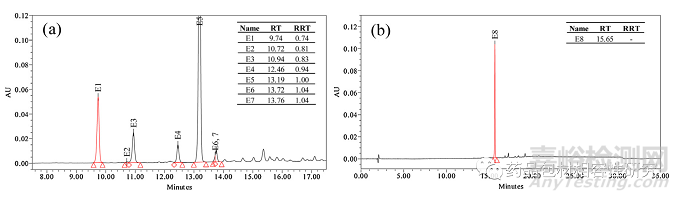
图3.代表性色谱图显示了来自品牌A(a)的可提取物E1-E7和来自品牌(b)的E8,它们用于鉴定。
3.5.1. E1 -E3 的识别
E1 的分子离子的精确质量数为 196.0413 Da,这可能对应于分 子式 C9H10NO2S+,根据元素计算器的精确质量数为 196.0427 Da。m/z 196 的 MS/MS 解离显示 m/z 178、168 和 150 的三个突出碎片(图 4一种)。m/z 178 和 168 的产物离子分别由 H2O 和 CO 的损失产生 (图 5a) 来自分子离子。从 m/z 168 的片段中进一步 丢失 H2O 或从 178 的片段中丢失 CO 导致 m/z 150 的片段。这 个后来的关键片段表明在氮上添加了羟乙基部分。它的紫外线解析(图S4)几乎与E3(核磁共振确认的结构)相似,这表明其基本 骨架与E3相似。基于这些观察和可能的分子式的最小误差 (-1.4 mmu),该可萃取物的结构可以建议为 3-(2-羟乙基)-2-苯并噻 唑啉酮。
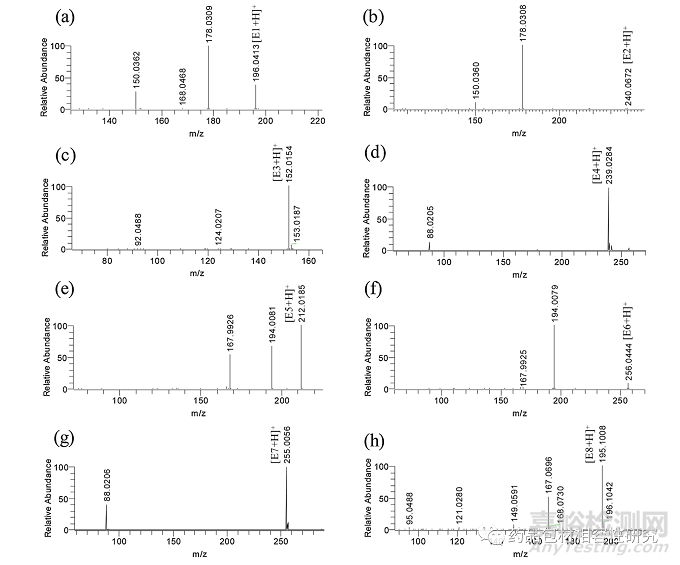
图 4. 来自品牌 A 和 B 注射器的可提取物的 LC-HRMS 线谱(分别为 E1-E8 的 a-h)。
E2 的质量为 240.0672 Da,比 E1 (图 4b) 并指出在 E1 的结构 中可能存在额外的羟乙基部分。它还在失去 C2H6O2时显示出 m/z 178(与 E1 相同)的碎片,在失去 CO 时进一步分解为 m/z 150 的离子。此外, E2 的 UV解析(图 S4)表明它的基本骨架类似于E1 和 E3,支持其提出的结构,即 3-(2-(2-羟基乙氧基)乙基)-2- 苯并噻唑啉酮 (图 5a).
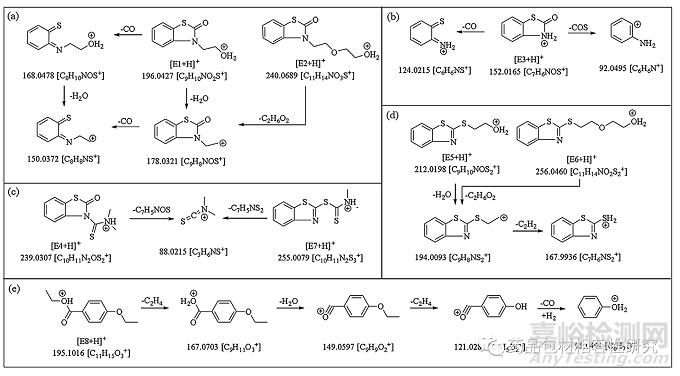
图5. E1和E2(a)、E3(b)、E4和E7(c)、E5和E6(d)和E8(e)的结构和质量碎裂行为
E3 显示 m/z 152.0154 (图 4C)。精确质量表明分子离子式为C7H6NOS+,精确质量为 152.0165。详细的 HRMS 数据以及 mmu 中 的质量误差包含在表格1。其分子离子表现出 CO 和 COS 的损失, 并分别产生 m/z 124 和 92 的产物离子(图 5b)。基于这些观察, E3 的结构被提议为 2-(3H)-苯并噻唑酮。
表格1. 来自品牌 A 和 B 注射器的可提取物的LC-HRMS数据。
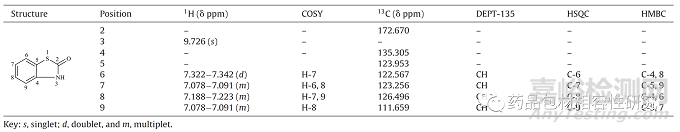
为了确认确切的结构,通过 LC-UV-MS(图 S5)对 E3 进行富集、分离和确认,以获得1H、13C 和 DEPT-135、COSY、HSQC 和 HMBC(图 S5)。S6-S10 分别和表(3) 核磁共振数据。1H NMR 谱(图 S6)显示化学位移值(δ ppm)处的信号:9.726 (s)、7.322-7.342 (d)、7.078-7.091 (m)、7.188-7.223 (m)和7.078-7.091 (m)的质子位置为3和6-9。δ9.726ppm 的化学位移值表明存在 NH 质子,而介于 δ7.0 至 δ7.4 ppm 之间的值证实了四个芳香族质子。13C NMR 光 谱显示在δ 172.670 、 δ135.305 、 δ123.953 、 δ122.567 、 δ123.256 、δ126.496 和δ 111.659 ppm 处的信号,对应于结构中的总共 7 个碳(图 S7)。DEPT-135 证实存在四个次甲基芳烃碳(图 S7)。位 于 δ135.305 和 δ123.953 ppm 的其余两个碳被指定为芳环的季 碳,它们在烯醇形式中可能更低,这表明 δ172.670 ppm 的信号可 能是羰基碳。IR数据(图S11)进一步支持了这一点,它清楚地显示 了1665.62 cm−1处的信号。2D 数据,即 COSY、HSQC 和 HMBC(分 别为图 S8-S10)进一步确定了所提出结构中的质子-质子和质子- 碳关系。基于以上数据,E3的结构被确认为2-(3H)-苯并噻唑酮。通过比较最近报道的同一化合物的 NMR 和 IR 数据进一步支持了 这种结构[19].
表3. E3 在 CD3 Cl 中的1H、COSY、13C、DEPT-135、HSQC 和 HMBC数据
对于与 E1 和 E3 相同的质量,Airaudo 等人 [16] 和Salmona 等人[17], 报道的结构为 2-(2-羟基乙氧基) 苯并噻唑 和 2-羟基苯并噻唑。虽然,我们的数据表明结构略有不同。在 E3 的情况下,酮不是烯醇形式,而是通过核磁共振、红外光谱和与 报告数据的比较来证明 [19] 而在 E1 中,基于 m/z 150 的关键 MS/MS 碎片以及 E1 和 E3 的紫外光谱之间的相似性,证明在 N 上添加羟乙基而不是 O 是合理的。
3.5.2. E4 和 E7 的识别
E4 和 E7 的准确质量数为 m/z 239.0284 (图 4d) 和 m/z 255.0056 (图 4g),分别具有与 E3 和 E5 几乎相似的紫外光谱(图 S4 ) 。它 们 的 分 子 式 被 鉴 定 为 C10H11N2OS2+(E4) 和 C10H11N2S3+(E7), 精确质量为分别为 m/z 239.0307 和 m/z 255.0079。从两个分子离子中看到的 m/z 88 的共同碎片表明存在 二甲基甲硫代酰胺阳离子(图 5c).
此提取物的形成已在下一节中解释。根据分子式,紫外光谱的相 似性和二甲基甲硫酰胺阳离子的常见碎片离子,E4和E7的结构被 提出为N,N-二甲基-2-氧代苯并[d]噻唑-3(2H)-碳硫酰胺和苯并[d] 噻唑2-基二甲基氨基二硫代酸酯,分别。最近,E7 的合成过程和 完整的表征数据已被报道[20].
3.5.3. E5 和 E6 的识别
E5 分子离子的准确质量为 212.0185 Da (图 4e)。可由此分子 量推导出的最接近的分子离子式确定为 C9H10NOS2+,精确质量为 212.0198 Da(误差 -1.3 mmu,对于所有其他可提取物遵循相同的 模式)。这显示了 m/z 194(H2O 丢失)和 168(C2H2丢失)的突 出片段,如图所示图 5d.根据 MS 数据和现有报告,它被鉴定为 2-(2-羟乙基巯基) 苯并噻唑 (HMBT),一种已知的可提取/可浸出 [16,17,21].
E6 的分子量为 m/z 256.0444 (图 4f),比 E5 的质量高 44Da。这种差异与在 E1 和 E2 之间观察到的相似。44 Da 的差异 表明 E5 中存在额外的羟乙基。m/z 194 和 168 的产物离子在 C2H6O2(如 E2 的情况)和 C2H2碎裂时的后续损失加强了其提出 的结构为 2- (2-(2-羟基乙氧基)乙基巯基)苯并噻唑(图 5d).
3.5.4. E8的识别
发现 E8 分子离子的准确质量为 195.1008 Da (图 4h),与最 近的分子式 C11H15O3+和精确质量 195.1016 Da(误差 -0.8mmu)。根据质量和分子式,可以将其鉴定为 4-乙氧基苯甲酸乙酯, 据报道该物质可从预装注射器中浸出[12].其 HRMS 谱显示 m/z 167、 149、121 和 95 的碎片,这些碎片可以在 C2H4、H2O、C2H4的亚 序列损失中产生和 CO (+H2) 来自分子离子 (图 5e),这进一步支 持了提议的结构。
3.6. 可提取物的建议途径
生成品牌 A 可提取物的暂定途径如图所示图 6。苯并噻唑衍生 物在橡胶生产中一般用作硫化促进剂[5,22,23]. 据报道,2-巯基苯并噻唑 (MBT) 在制造一次性注射器的垫圈时被用作促进剂或苯并噻唑衍生物,在硫化过程中会转化为它[6,15–17,21]。已知其 与杀菌剂即环氧乙烷反应生成HMBT即E5[21,24]. MBT 和 HMBT 对苯 并噻唑 2 位羟基离子的攻击可生成 E3,即 2(3H)-苯并噻唑酮 [24]. 环氧乙烷与 E3 的 NH 质子反应可能导致形成 E1,即 3-(2- 羟乙基)-2-苯并噻唑啉酮 [24]. 在 E1 和 E5 的游离羟基上进一步 加成环氧乙烷可分别导致 E2 和 E6 的形成。E4 和 E7 可以通过 二级促进剂如四甲基秋兰姆单硫化物 (TMTM) 和四甲基秋兰姆二硫 化物 (TMTD) 的 N, N-二甲基甲硫代酰胺部分的反应生成。20,25,26] 分别使用 E3 和 MBT/HMBT。已知 E8,即 4-乙氧基苯甲 酸乙酯可从聚乙烯或聚丙烯制成的塑料中提取出来,可用作聚合 催化剂[12,27–29]. 在这种情况下,大部分从橡胶垫圈中流出(尽管在塑料桶的情况下检测到少量,但在 温和的提取条件下),它可以用作聚合催化剂。所有八种确定的可提 取物的摘要提供在表 4。
4. 结论
该研究涉及在研究药物产品的 LC 方法开发过程中观察到与已知 杂质共流出的可浸出物后,对一次性临床级塑料注射器的可提取物进 行全面调查。观察到的可提取物数量和类型的差异强调了来自不同品 牌注射器的潜在可浸出物含量的差异。在 LC-HRMS 和/或 NMR/IR 的 帮助下,鉴定了塑料一次性注射器中的八种可提取物,并提出了其形成的合理途径。据我们所知,四个提议的可提取物尚未报告为可提取/可浸出物,而三个被发现具有化学新颖性。这些发现在常规药品分析中特别重要,其中分析方法可能对这些可提取物没有选择性,因此在纯度/杂质测定过程中构成风险。这项工作还证明了仅用注射用水提取临床级注射器中的可浸出物, 这可能是一个潜在的安全问题,值得进一步调查。

来源:Internet


