您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2019-10-24 09:05
01 引言
通过510(k)获得FDA认证,是医疗器械进入美国市场最常见的申请途径。根据不完全统计,2018年FDA共批复约3000件510(k)申请。为了让审核更加高效,节省企业资源,FDA在今年9月份出台了一份指南文件“传统和简化版510(k)的格式”,用来指导企业编写510(k)的申请材料。今天,小编就带大家来看看这份最新的文件清单。
02 传统和简化版510(k)文件清单
指南文件建议厂商在提交510(k)申请资料时,按照下表进行汇编,这份清单总共包括20份文件。考虑到并不是每份文件都适用于所有的医疗器械,对于有些不适用的情况,指南文件建议厂商不要删除,只需要在文件名中加上“This section does not apply”或者“N/A”即可。
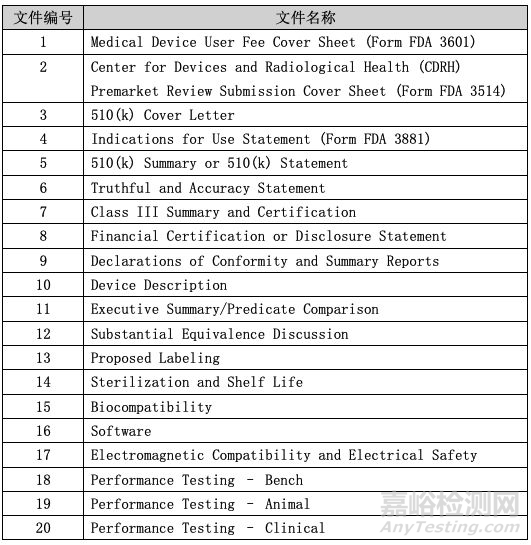
03 每份文件的具体描述
1. Medical Device User Fee Cover Sheet
即用户费用付款收据或有效的电子副本,以允许FDA开始处理厂商提交的申请。
2. CDRH Premarket Review Submission Cover Sheet
该文件属于自愿性表格,用于提供申请的基本行政信息。
3. 510(k) Cover Letter
即510(k)附函,其目的是为510(k)的初步处理和审核提供关键信息,保证审核的有效性。根据指南文件附录A,附函可以包括以下内容:510(k)类型、器械类别、提交人、主要通讯者、保密性、分类法规号、具体分类、大类、产品代码、与器械相关的FDA文件编号、提交传统或简化510(k)申请的依据、以及器械设计和使用。
4. Indications for Use Statement
即适应症说明,特别要明确器械是处方使用还是非处方使用,建议使用FDA表格3881.12。
5. 510(k) Summary or 510(k) Statement
510(k)总结包含器械的简要介绍和支持性信息;510(k)声明是对持有人将在书面请求后30天内,向任何人提供510(k)申请副本的证明。
6. Truthful and Accuracy Statement
即真实性和准确性声明,证明在510(k)中提交的所有信息都是真实准确的,并且没有遗漏任何重要事实。510(k)持有人应在真实和准确声明上签名并注明日期。
7. Class III Summary and Certification
如果510(k)适用于未要求PMA且归类为III类的器械,则必须包含总结和符合21 CFR 807.87(k)和807.94的证明。其中,“III类器械总结”要对与类比器械相关的安全性和有效性问题的类别进行总结,并引用参考信息。“证明” 则确保对所有相关III类器械和其他类似合法销售器械的已知或其它可用信息进行合理检索。
8. Financial Certification or Disclosure Statement
根据21 CFR 807.87(i),如果提交来自临床研究的信息,则必须为参加研究的每个临床研究者提交财务证明或披露声明。
9. Declarations of Conformity and Summary Reports
即符合声明和和总结报告。如果是申请传统510(k),则提供使用非强制性标准的相关信息,包括任何符合性声明或此类标准的使用依据;如果是申请简化510(k),则提供使用标准的相关信息或总结报告,该报告用于描述如何满足特定器械指南中的特殊要求。
10. Device Description
在器械描述部分,建议包括性能参数、器械设计要求、型号、和附件的描述。如果有助于理解的话,可提供图表、尺寸、偏差、或原理图,同时也建议提供所有与人体接触的成分及其组成材料的清单。
11. Executive Summary/Predicate Comparison
概要/对比表格部分主要会包含:器械的简洁描述(包括适应症和技术)、器械对比表格、以及提交的任何性能测试的概述。指南文件建议除了绘制对比表格外,还要提供实质等同的证据,同时性能测试总结应该包括测试方法、结果和结论。
12. Substantial Equivalence Discussion
实质等同讨论部分,建议明确对比器械的商品名、型号、510(k)申请人、510(k)号,并且从适应症、技术和性能参数(包括任何测试)方面做详细对比。
13. Proposed Labeling
510(k)必须包括足够详细的标签信息,以满足21 CFR 807.87(e)的要求。如果是体外诊断器械,则标签必须满足21 CFR 809.10的要求。标签通常会包括器械标签、使用说明书以及任何患者标签。
14. Sterilization and Shelf Life
对于无菌器械,应参考指南“Submission and Review of Sterility Information in Premarket Notification (510(k)) Submissions for Devices Labeled as Sterile”。
对于再处理的一次性医疗器械,应参考指南“Medical Device User Fee and Modernization Act of 2002 Validation Data in Premarket Notification Submissions (510(k)s) for Reprocessed Single-Use Medical Devices”。
申请中宣称的器械效期,应该通过实验室测试以及灭菌和包装确认来证明。
15. Biocompatibility
如果器械组成直接或间接与人体接触,则需要评估与人体接触材料的生物相容性。可参考FDA指南文件“Use of International Standard ISO 10993-1, "Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process”。
16. Software
软件描述文档可参考指南“Guidance for the Content of Premarket Submissions for Software Contained in Medical Devices”;网络安全信息描述参考指南“Content of Premarket Submissions for Management of Cybersecurity in Medical Devices”。
17. Electromagnetic Compatibility and Electrical Safety
如果器械通过电驱动,则需要评估电磁兼容性(EMC),包括辐射和抗干扰,参考“Information to Support a Claim of Electromagnetic Compatibility (EMC) of Electrically-Powered Medical Devices”。
电器安全则参考:ANSI/AAMI ES 60601-1: Medical electrical equipment - Part 1: General requirements for basic safety and essential performance or an equivalent method.
18. Performance Testing–Bench
性能测试信息描述可以参考指南“Recommended Content and Format of Non-Clinical Bench Performance Testing Information in Premarket Submissions.”
19. Performance Testing–Animal
关于动物测试,建议描述测试方法并提供证明器械性能的测试结果。通常会提交以下信息:罗列实施的具体动物试验、描述测试方案、结果、分析、以及讨论。
测试方案需包括:测试目的、测试中使用的测试主体、测试方法和过程(包括任何特定测试条件)、研究终点和通过/失败接受标准。
另外,指南文件建议在结果和分析中,以简洁明了的形式(例如表格)介绍测试数据,并在结论中从实质等效性的角度描述与对比器械的比较测试。
20. Performance Testing–Clinical
提供的临床方案应明确测试目的、测试方法和过程(包括特定测试条件)、研究终点(包括安全性和有效性)、以及统计学方法。此外,还要讨论研究结果、分析和结论,并讨论与对比器械的实质等同。
对于开展的临床试验,需要满足ClinicalTrials.gov的要求并获得合规证明。如果是高风险器械,则需满足21 CFR Part 812;如果是低风险器械,则满足21 CFR Part 812.2(b)。
对于提交给FDA的海外临床试验数据,需满足21 CFR 812.28的要求。

来源:启升资讯


