FDA官网中一个有关药物开发报告的实例,用以说明申请人如何实施质量源于设计(QbD)。 该实例的目的是说明ANDA申请人在其仿制药开发过程中实施QbD时,可使用的药物开发研究的类型,同时促进探讨OGD在审评中如何使用该信息。
本文主要概述了制剂处方开发的变更风险关键要点和处方开发的案例1。
2.2 Drug Product 制剂
2.2.1 Formulation Development 处方开发
2.2.1.1 Initial Risk Assessment of the Formulation Variables 处方变量的初始风险评估
在该处方开发的初始风险评估中,还未建立详细的生产工艺。因此,假定对于每个 变更的处方属性,将建立优化的生产工艺来评估风险。
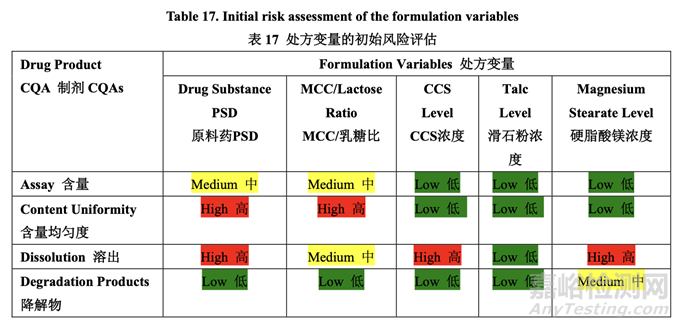
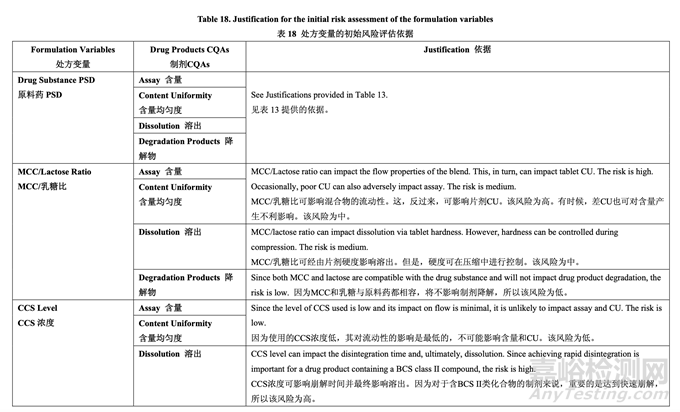
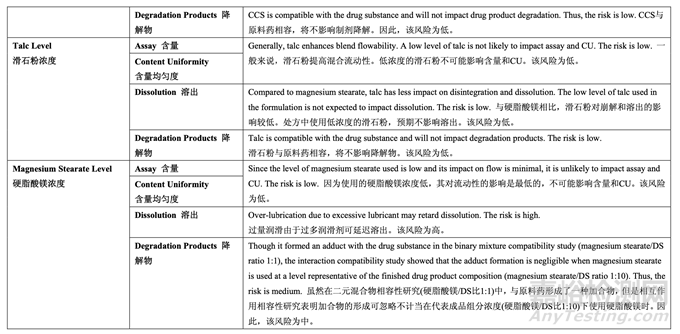
处方变量的初始风险评估结果见表 17,风险分配的依据见表 18。
2.2.1.2 Drug Substance Particle Size Selection for Product Development
用于产品开发的原料药粒径选择
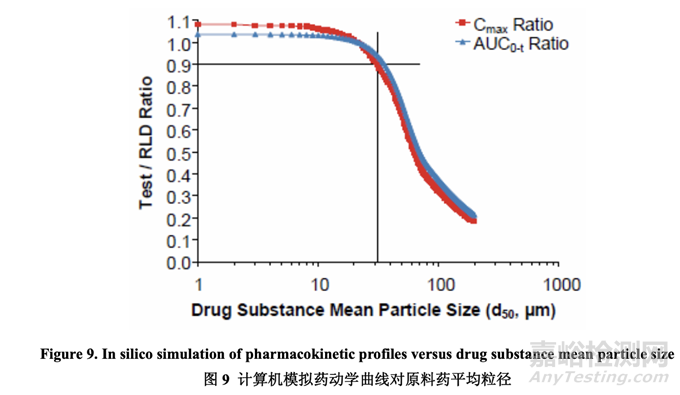
一般来说,对于板状形态和粒径在微米范围内的原料药,较大粒径的原料药可提高生产性因 为其流动性较好。但是,对于像 acetriptan 的 BCS II 类化合物,较大粒径的原料药可显著减 少溶出,对体内性能产生消极影响。为确定用于进一步研究的适宜原料药粒度分布范围,进 行了计算机模拟以估计原料药平均粒径 d50 对受试制剂和 RLD 间的 Cmax 比和 AUC 比的影 响。5 预定义的选择标准是产生 Cmax 比和 AUC 比为 0.9~1.11 的平均粒径。d50 范围从 1 μm 至 200 μm 的模拟结果以图表表示,见图 9。数据表明 d50 为 30 μm 或以下符合预定义标准, 显示出对药动学曲线有限的影响当与 RLD 比较时。
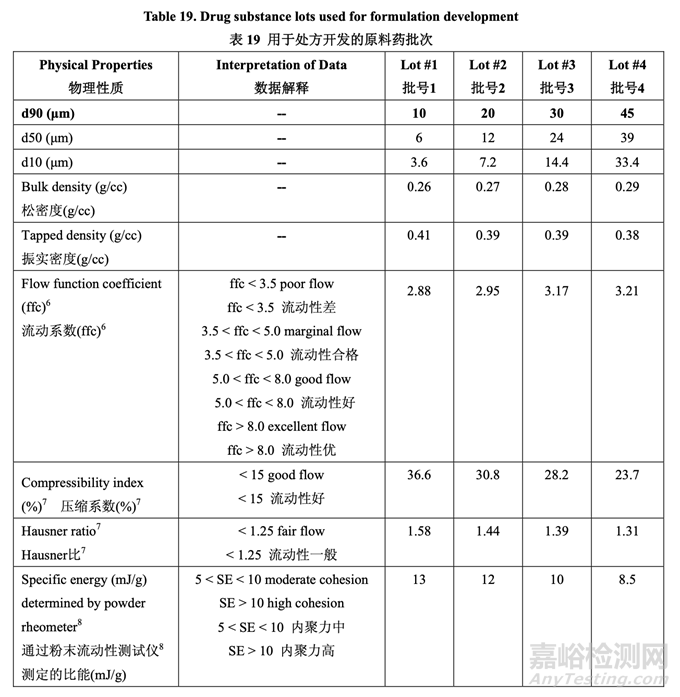
基于模拟结果,选择不同粒度分布的原料药 4 个批次用于处方开发。最终的目的是测试中试 PK 研究中的处方以最终确定用于工业化的原料药粒度分布。评估了原料药 4 个批次的物理 性质和流动性并总结在表 19 中。在该开发报告中,d90 用于描述原料药粒度分布。Acetriptan 的d90分别为10 μm, 20 μm, 30 μm和45 μm分别对应于d50为6 μm, 12 μm, 24 μm和39 μm。
2.2.1.3 Process Selection 工艺选择
当d90在10~45 μm范围内时,acetriptan具有内聚性并显示出差流动性,如压缩系数,Hausner 比,流动系数和比能所表明。物料的流动性差可产生具有重量差异和含量差异高的片剂,由 于原料药在混合物中的不均匀分布,不均匀松密度并最终导致压片机上的膜腔填料不均匀。
Acetriptan差流动性排除了处方高载药量的可能性,支持使用与RLD类似的载药量(10%)。 Initially, direct compression of the blend was performed. The blend uniformity (BU) percent relative standard deviation (% RSD) was higher than 6% and the tablet content uniformity % RSD was even higher. Therefore, direct compression was considered an unacceptable process for this formulation.
开始,对混合物进行了直压。混合物均匀度(BU)相对标准偏差百分率(% RSD)高于6,片剂 含量均匀度% RSD更高。因此,该处方的直压工艺被认为是不可接受的。
排除了湿法制粒由于干燥中原料药的潜在热降解,基于强降解研究结果。也排除了使用有机 溶剂的湿法制粒因为要求避免涉及的环保考虑。对于碾压干法制粒,原料药的粉末颗粒和填 充剂在高压下聚集形成带状物,然后在压缩(压片)前,通过粉碎分解产生颗粒。可最大限度 减少药物颗粒分离的风险。通过控制颗粒的粒度分布和流动性,可降低片剂含量均匀度差的 风险。因此,选择碾压干法制粒作为制剂进一步开发计划的工艺。
2.2.1.4 Formulation Development Study #1 处方开发研究#1
在因素间无潜在相互作用的情况下,单变量法(即单次单因素(OFAT))是可接受的。 因为这经常是未知的,所以经常使用多元统计设计(即实验设计(DOE))并用市售可用的统计 软件评估结果。当计划DOE时,普遍使用序贯策略。起初,筛选DOE可用于缩小初始风险 评估中确定为一些关键因素的大量的因素名单。然后,表征DOE可用于理解主要作用和这些 关键因素间的潜在相互作用。当中心点包含在2水平析因DOE中时,检验曲率效应是否显著 是可能的。通过从校正模型的回归模型中分离出曲率项来进行数据分析。如果曲率显著,则 应增加至反应面DOE设计以估计二次项。另一方面,如果曲率不显著,则校正模型和不校正 模型是类似的。最后,经验证的DOE可用于研究系统的耐用性,通过预期在常规生产中遇到 的范围内,变更已确定的关键因素。
随机化,分组和重复是统计实验设计的3个基本原则。通过对实验进行适宜随机化,可能存 在的不可控因素可“达到平均数”。分组是将试验单位分为彼此类似的群(组)。分组降低了 差异在群间的已知但无关来源,因此在研究中估计差异来源的精密度更高。重复可估计纯试 验误差以确定数据中观察到的差异是否具有真实统计学差异。
在该模拟例子中,我们未包含每个DOE的ANOVA结果。实际上,请注意ANOVA结果应附有 所有的DOE数据分析,特别是如果讨论了关于模型项意义的结论。对于所有的DOE数据分析,选择常用的α=0.05以区分显著因素和非显著因素。
重要的是任何实验设计具有足够的功效以保证得出的结论是有意义的。可通过计算信噪比来 估计功效。如果功效低于预期水平,则某些补救措施可用于增加功效,例如,通过加入更多 批次,增加信号或减少系统噪音。请参阅 ICH 要点考虑文件,关于药政提交建议的 DOE 记 录水平的指南。
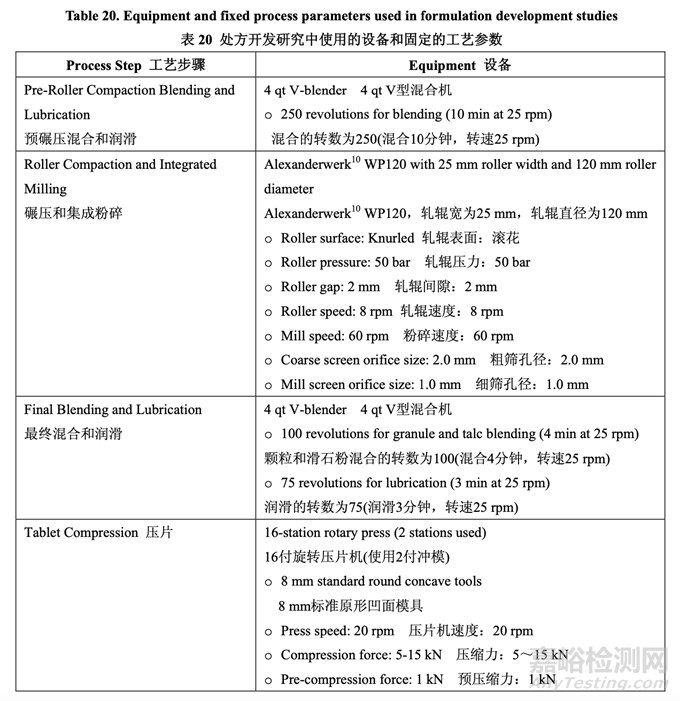
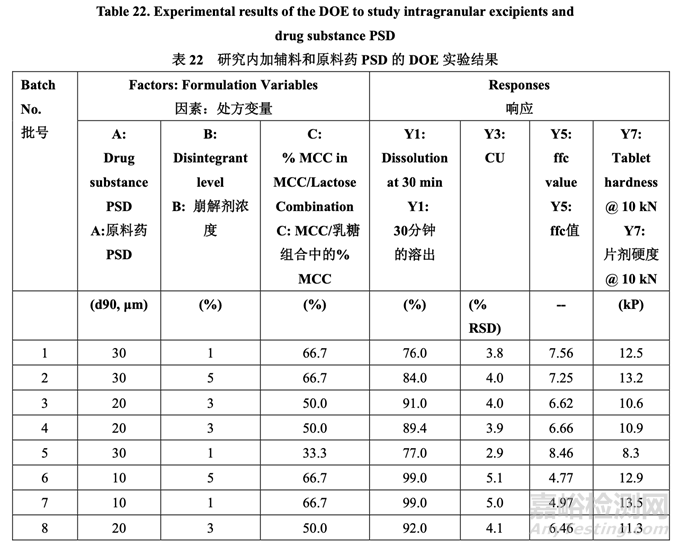
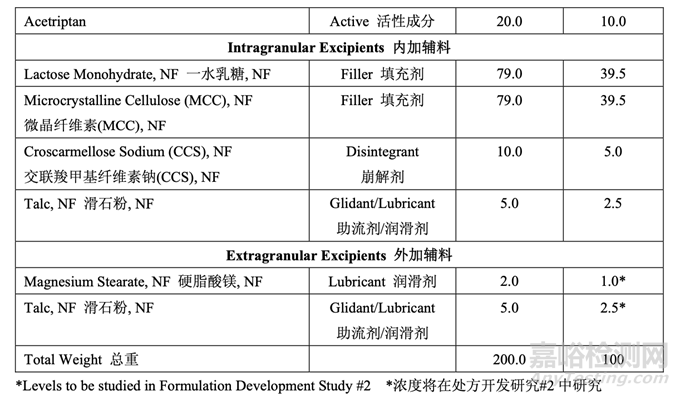
处方开发集中于初始风险评估中确定的高风险处方变量的评估,如表 17 所示。开发在 2 个 阶段进行。第一个处方研究评估了原料药粒度分布,MCC/乳糖比和崩解剂浓度对制剂 CQAs 的影响。进行第二个处方研究以理解处方中外加硬脂酸镁和滑石粉浓度对产品质量和生产性 的影响。在实验室规模(1.0 kg, 5000 单位)下进行了处方开发研究。表 20 详细说明了这些研 究中使用的设备和相关的工艺参数。
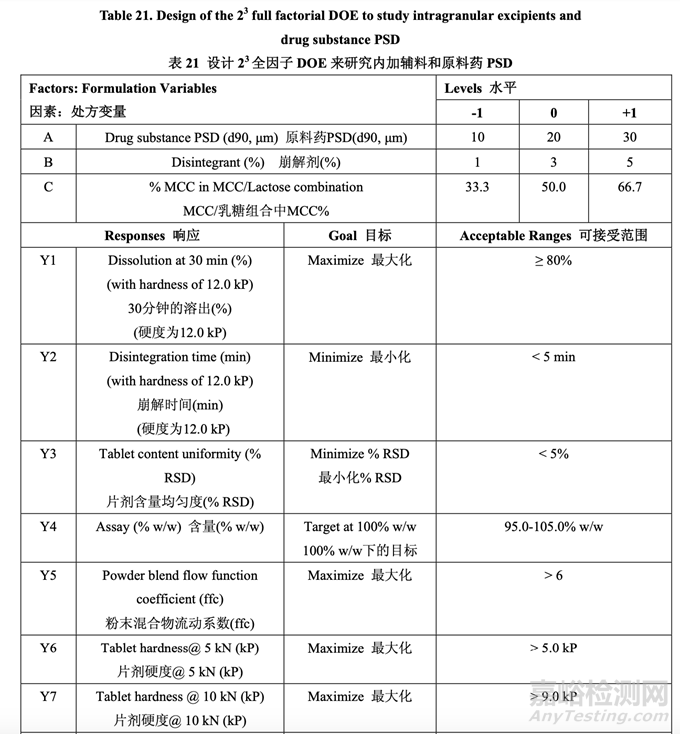
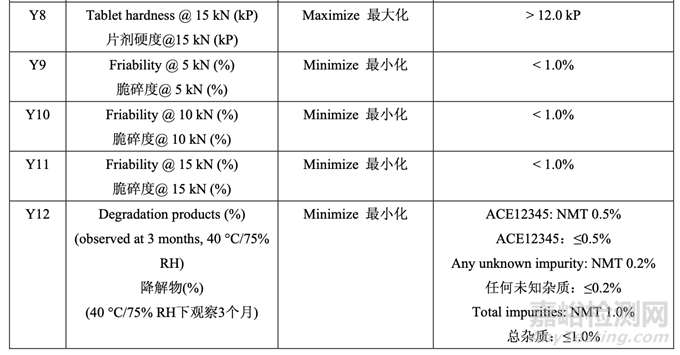
处方开发研究#1的目的是选择MCC/乳糖比和崩解剂浓度并理解这些变量是否与原料药粒度 分布存在相互作用。该研究也试图建立拟定处方的耐用性。3中心点的23全因子实验设计 (DOE)用于研究这3个处方因素对表21所列响应变量的影响。
这些原料药 批的特征见表 19 并基于 2.2.1.2 节讨论的计算机模拟结果而进行选择。
以内加方式加入崩解剂(交联羧甲基纤维素钠),研究的浓度范围从 1%至 5%。这些浓度与RLD 处方中估计的浓度一致,都在药用辅料手册推荐的范围内。11
选择用于处方研究的MCC/乳糖比是基于之前批准的使用碾压生产的产品(ANDA 123456和 ANDA 456123)的经验。通过分配值为33.3%, 50.0%和66.7%分别对应于1:2, 1:1和2:1,MCC/ 乳糖比随MCC/乳糖二元填充剂组合中MCC百分比而转换为连续数字变量。
仿制药处方的载药量固定为 10%,基于 RLD 标签,规格和片重。对于该研究,内加和外加 滑石粉浓度都固定为2.5%。外加硬脂酸镁浓度固定为1%。滑石粉和硬脂酸镁浓度都与RLD 处方中观察到的浓度一致并与药用辅料手册公布的建议一致。 使用的片重恒定为 200.0 mg,调整填充剂数量以达到目标重量。
表 21 总结了研究的因素和响应。对于每批,以几种压缩力(仅显示 5 kN, 10 kN 和 15 kN 的数 据)压缩混合物以得到压缩曲线。使用该曲线,调整压缩力来压缩片剂达到崩解和溶出检查 的目标硬度。
为研究片剂目标硬度 12.0 kP (允许的范围为 11.0~13.0 kP)下的片剂溶出,调整了压缩力。 选择片剂目标硬度 12.0 kP 以研究处方变量对溶出的影响因为高硬度预计是溶出的最坏情 况。如果在固定压缩力下研究溶出,则结果可能受片剂硬度影响的混淆。
在碾压(Y6)前,使用环剪仪测量粉末的流动系数(ffc)。根据文献,以下规则用于判断粉末的 相对流动性:
ffc<3.5poor ffc<3.5 差
3.5<ffc<5.0marginal 5.0<ffc<8.0good
ffc > 8.0 excellent
3.5<ffc<5.0 合格 5.0<ffc<8.0 好
ffc > 8.0 优
以 10 kN (Y1, Y3, Y5 和 Y7,其他响应未显示)压缩的溶出,含量均匀度,粉末混合物流动系数 和片剂硬度的实验结果见表 22。
Significant factors for tablet dissolution (at 30 min) 片剂溶出(30分钟)的显著因素
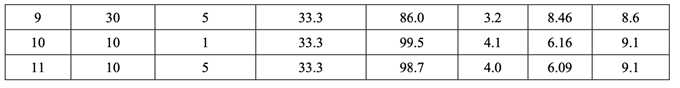
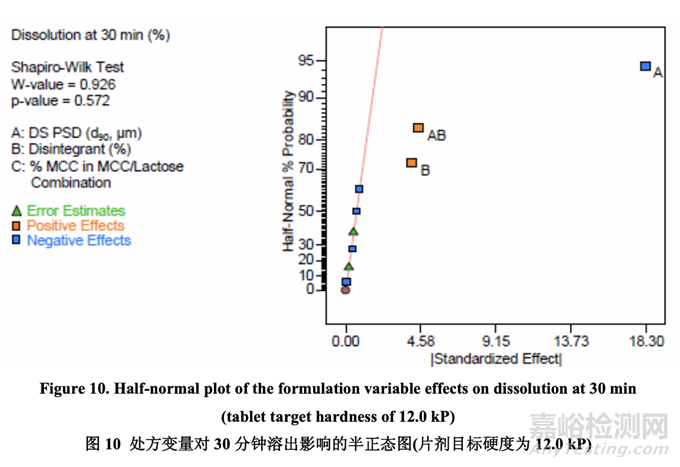
最初,使用 FDA 推荐的方法检查溶出。所有批显示出快速和类似于 RLD 的溶出(30 分钟内 溶出> 90%)。然后使用内部溶出方法再次检查所有批(具体见 1.4 节)。结果见表 22。因为中 心点包括在 DOE 中,所以使用校正模型检验曲率效应的意义。方差分析(ANOVA)结果见表 23。
如表 23 所示,溶出的曲率效应无意义;因此,使用所有数据(包括中心点)来拟合因素模型 系数。如以下半正态图(图 10)和未校正模型的 ANOVA 结果(表 24)所示,影响片剂溶出的显 著因素是 A (原料药 PSD), B (崩解剂浓度)和 AB (原料药 PSD 和内加崩解剂浓度间的相互作用)。
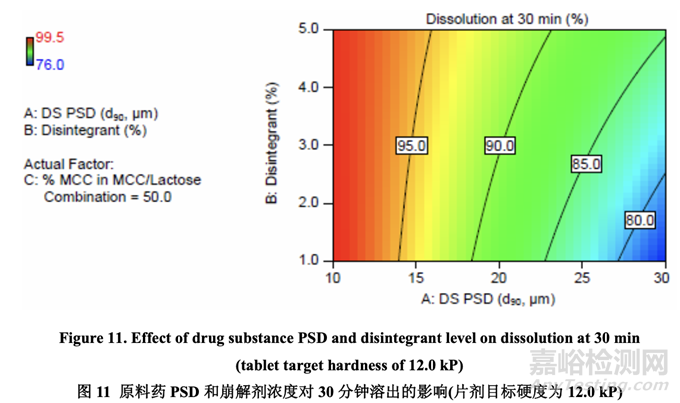
图 11 显示了原料药 PSD 和崩解剂浓度对 30 分钟时溶出的影响。溶出随原料药 PSD 增加而 减少。另一方面,溶出随崩解剂浓度增加而增加。用较大的原料药 PSD,崩解剂浓度对溶 出的影响大于用较小的原料药 PSD。
Significant factors for tablet disintegration time 片剂崩解时间的显著因素
崩解剂浓度是影响片剂崩解的唯一具有统计学意义的因素。但是,所有批显示在不到4分钟 内快速崩解。
Significant factors for tablet assay 片剂含量的显著因素
所有批显示出可接受含量(范围为98.3~101.2%),位于质量标准限度内(95.0~105.0% w/w), 无因素显示出对片剂含量具有显著影响。
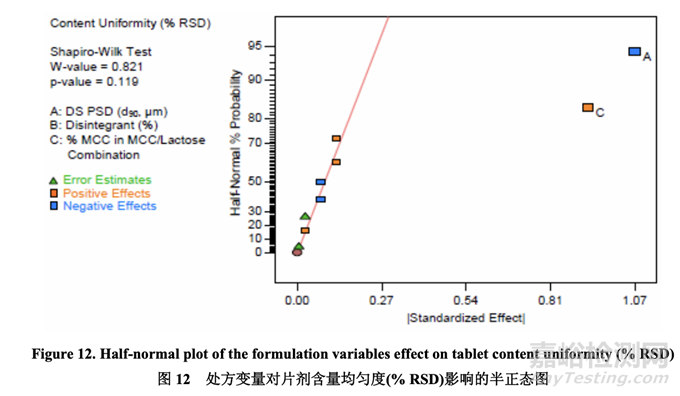
Significant factors for tablet content uniformity (%RSD) 片剂含量均匀度(%RSD)的显著因素
数据分析表明曲率效应对于片剂含量均匀度不显著。如半正态图(图 12)所示,影响片剂含量 均匀度的显著因素是 A (原料药 PSD) 和 C (MCC/乳糖组合中的% MCC)。
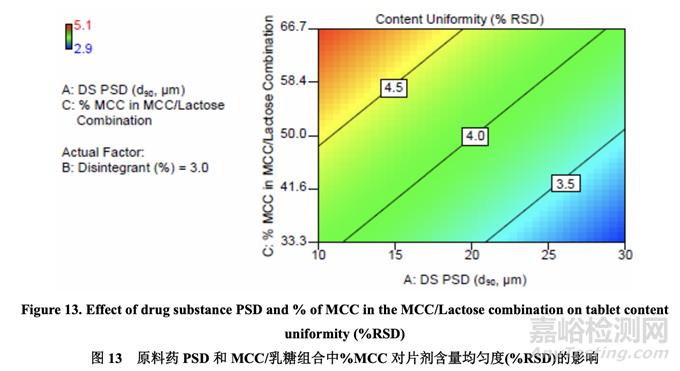
图13显示了原料药PSD和MCC/乳糖组合中的% MCC对片剂含量均匀度的影响。% RSD 随原料药 PSD 增加而减少。另一方面,% RSD 随 MCC/乳糖组合中的% MCC 增加而增加, 可能是因为 MCC 的纤维颗粒形状与乳糖的球形颗粒形状一样不具有流动性。
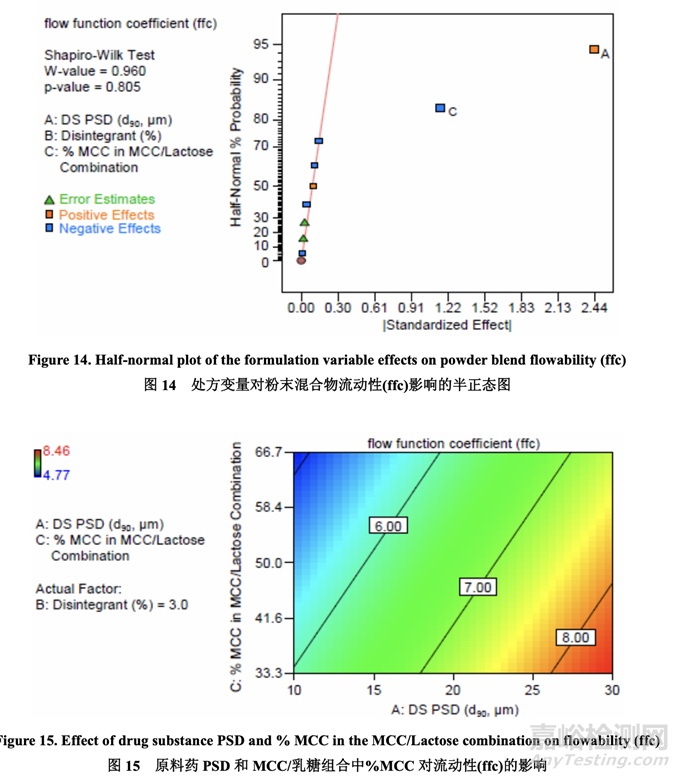
Significant factors for powder blend flowability 粉末混合物流动性的显著因素
使用环剪仪测定每个样品的预碾压混合和润滑步骤中粉末混合物的流动性(以 ffc 值表示)。 然后记录每个样品的 ffc。如半正态图(图 14)所示,影响粉末混合物流动性的显著因素是 A (原 料药 PSD) 和 C (MCC/乳糖组合中的% MCC)。原料药 PSD 和 MCC/乳糖组合中的% MCC 对粉末混合物流动性的影响如图 15 所示。粉末混合物流动性随原料药 PSD 增加而增加,随 MCC/乳糖组合中的% MCC 减少而增加。
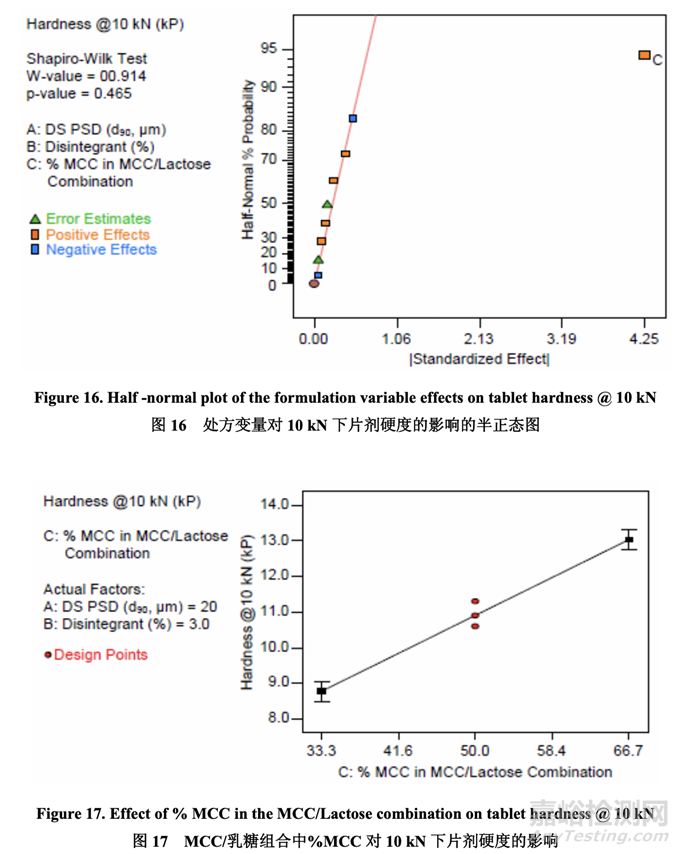
Significant factors for tablet hardness 片剂硬度的显著因素
每个 DOE 批在 5 kN, 10 kN 和 15 kN 下压制以评估其可压性。半正态图(图 16)显示,当使用 10 kN 压缩力时,唯一影响片剂硬度的显著因素是 C (MCC/乳糖组合中的% MCC)。压缩力 为 5 kN 和 15 kN 可观察到类似的关系(数据未显示)。如图 17 所示,在给定压缩力下,片剂 硬度随 MCC/乳糖组合中的% MCC 增加而增加。
Significant factors for tablet friability 片剂脆碎度的显著因素
在5 kN, 10 kN和15 kN下压制的所有片剂显示出良好的脆碎度(片剂硬度范围为5.0~12.0 kP 的重量损失< 0.2%),研究范围内的3个处方变量未显示出对片剂脆碎度有统计学意义的影 响。
Significant factors for tablet stability (degradation products) 片剂稳定性(降解物)的显著因素
40 °C/75% RH下,将所有实验批置于稳定箱中的敞口容器中3个月,定期取样并分析。降解 物ACE 12345,单个未知杂质和总杂质都分别低于质量标准限度0.5%, 0.2%和1.0%。处方变 量未显示出对降解物有统计学意义的影响。
Summary of Formulation Development Study #1 处方开发研究#1的总结
Acetriptan PSD对片剂溶出,含量均匀度和粉末混合物流动性有显著影响。原料药PSD较小 可提高溶出;但是,它对片剂含量均匀度和混合物流动性产生消极影响。
内加崩解剂浓度显示出对片剂溶出有显著影响,由于其与原料药PSD的相互作用。崩解剂浓 度对溶出具有较大的影响当原料药PSD较大时。
MCC/乳糖组合中的% MCC对粉末混合物流动性,片剂含量均匀度和片剂硬度有显著影响。 增加% MCC可增加片剂硬度但减少了粉末混合物流动性并对片剂含量均匀度产生消极影 响,证据是% RSD增加了。为平衡混合物流动性和片剂硬度,选择用于暂定的处方为MCC/ 乳糖组合中的50% MCC(即1:1比)。
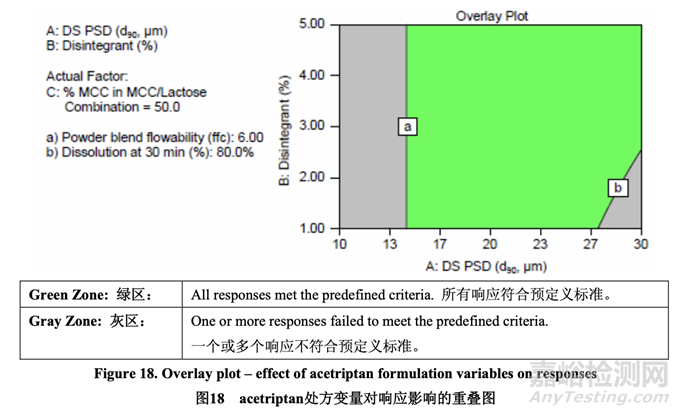
因为研究的任何响应未观察到曲率效应,以及使用无重叠项的全因子DOE确定了主效应和交 换效应,所以无需进一步研究来优化内加辅料。DOE模型用于建立处方变量的可接受范围。 图18显示了所有响应的重叠图。绿区表明所有响应都同时实现了。
为容纳可能最大的原料药PSD并避免在溶出可能不合格的绿区边缘操作,选择5%交联羧甲 基纤维素钠用于暂定处方。用所选的该崩解剂浓度,原料药d90的可接受范围为14~30 μm。 d90低于14 μm显示出流动性不适宜,导致片剂的含量均匀度不合格,当在处方开发中使用固 定的生产工艺时。因此,在预碾压混合和润滑工艺开发中进一步研究了原料药PSD。
为理解原料药PSD对体内性能的影响并确定仍可能生物等效的粒径上限,中试BE研究中研 究了d90为20 μm, 30 μm和45 μm(相当于d50分别为12 μm, 24 μm和39 μm)的原料药(见1.4节)。
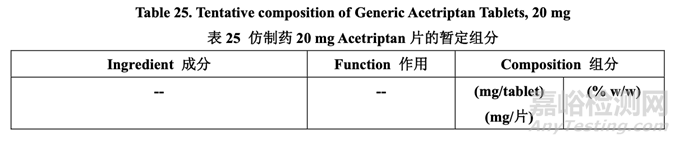
在处方开发研究#1 结束时,暂定了内加辅料的浓度,如表 25 所示。处方开发研究#2 进一步 研究了外加助流剂和润滑剂。
参考文献:
Example QbD IR Tablet Module 3 Quality 3.2.P.2 Pharmaceutical Development,FDA,2012.