前言
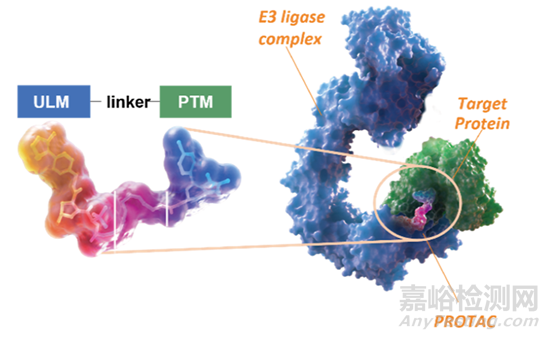
蛋白降解靶向嵌合体(即Proteolysis-Targeting Chimeras,在上下文中简称为“PROTAC”)是由靶向蛋白的配体(protein targeting moiety, PTM)与泛素连接酶配体(ubiquitin ligase moiety, ULM)连接而成的异双功能分子(图1)。它们的功能是诱导靶蛋白与泛素连接酶接近,导致泛素转移并随后引起泛素蛋白酶体系统对靶蛋白的降解。在过去的十年中,PROTAC作为新型治疗药物在学术界和制药行业引发的关注呈现爆炸式增长。
图1. PROTAC与靶蛋白之间诱导的三聚体复合物的结构-PROTAC-E3连接酶
与此同时,开发口服PROTAC药物的最大障碍,是实现有效的口服吸收。就其性质而言,PROTAC不同于传统的小分子药物,不满足经典的类药五原则(Rule of Five),但却在临床研究中被证明有口服吸收的潜力。最近,人们对扩展五原则(eRo5)或者超越五原则(bRo5)的分子重新产生了兴趣,Keith R. Hornberger和 Erika M. V. Araujo在Journal of Medicinal Chemistry杂志上发表了一篇文章,标题为Physicochemical Property Determinants of Oral Absorption for PROTAC Protein Degraders。在这篇文章中,他们利用ARVINAS公司现有的PROTAC数据库,对影响PROTAC口服吸收的物理化学性质因素进行了全面的分析,讨论了PROTAC在eRo5/bRo5空间中的位置,并针对口服PROTAC药物的设计得出了几个关键性结论。本文旨在对该文章进行详细的解读,希望为PROTAC药物的研发提供思路。
引入评价吸收的关键指标摒利用度指标弃常用的口服生物
口服生物利用度(F)的定义为F = fa × fg × fh,其中fa为吸收分数,fg为胃肠道有效通过分数,fh为肝有效通过分数。口服吸收药物经肝门静脉进入肝脏,再经肝脏首过代谢后进入体循环, fh 可以定义为 1 -(Cl/Qh),其中Cl为静脉给药的体内清除率,Qh为肝血流量。口服生物利用度Fobs可在实验中通过对比口服和静脉给药后的血药浓度时间曲线下面积(AUC)得到,但fa × fg从定义上更能代表药物从肠道吸收的情况。虽然很难通过实验直接测定fa或fg的值,但两者的乘积可以估算,即fa × fg = Fobs /(1- (Cl/Qh ))。
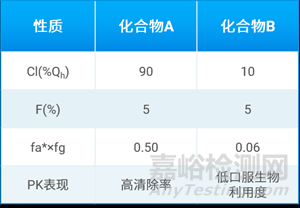
许多先前将分子的物理化学性质与口服生物利用度Fobs联系起来进行分析的方法是有一定缺陷的。为了说明这一点,Keith R. Hornberger和 Erika M. V. Araujo分析了表1中两个化合物的情况。
表1. 口服生物利用度和肠道吸收情况存在潜在不一致
从表中可以看到,尽管两个化合物的口服生物利用度均为5%,但是两个化合物的肠道吸收能力差异较大。化合物A具有较高的肝脏清除率(90%肝血流量),而fa × fg为0.5,高肝脏清除是造成低口服生物利用度的主要因素。化合物B的肝脏清除率相对比较低(10%肝血流量),而fa × fg仅为0.06,低的肠道吸收是造成低口服生物利用度的主要因素。因此,fa × fg相比Fobs更适用于评估化合物的肠道吸收情况,它将肝脏清除率因素对生物利用度影响排除在外。
根据经验设定fa × fg的阈值为0.25
通常认为一个比较令人满意的吸收情况是化合物至少有20%的口服生物利用度和比较低的肝脏清除率(<25% Qh),计算得出的最小fa × fg约为0.26。因此,在后续的统计中,将fa × fg = 0.25设定为口服PROTAC分子一个阈值。
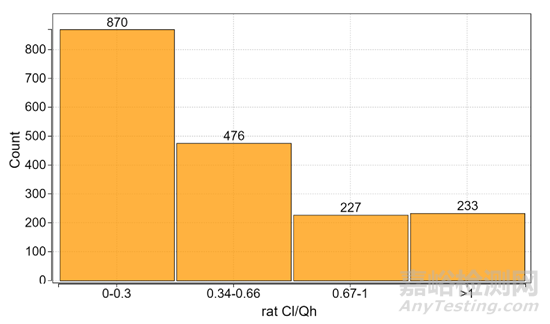
本次共有1806个PROTAC化合物参与了统计,选择的种属是大鼠,主要因为大鼠的静脉和口服PK数据比较全面,同时大鼠也是临床前毒理实验常用的种属。首先对这1806个化合物的fa × fg值进行了计算和统计,从图2的结果中可以看到75%的化合物显示了低或中等的体内清除,证明清除率不是影响口服吸收的主要因素。另外有233个化合物(约占13%)的清除率大于大鼠肝血流量,因此在后续的统计中剔除了这些清除率大于Qh(55 mL/min/kg)的分子,否则使用公式fa × fg = Fobs /(1- (Cl/Qh ))无法对数据进行有意义的评估。
图2. 大鼠中Cl/Qh 值的分布(n=1806)
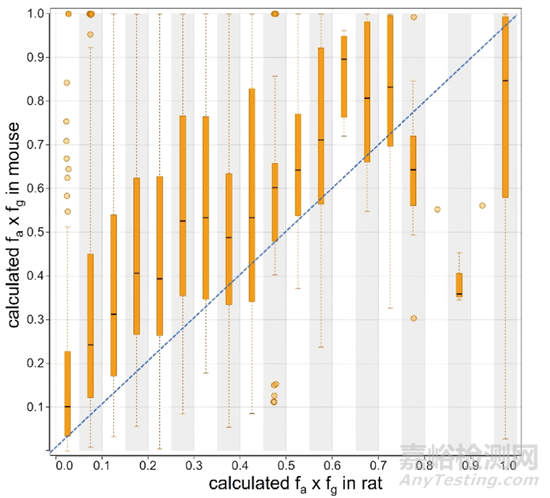
在对剩余的1573个化合物进行分析之前,首先对能同时得到大鼠和小鼠PK数据的1016个化合物进行了分析,利用小鼠的肝血流量90 mL/min/kg和大鼠的肝血流量55 mL/min/kg分别进行fa × fg计算,从图3的结果中可以看到85%(861/1016)的PROTAC分子在大鼠中的fa × fg是低于在小鼠中的,即大多数PROTAC分子在大鼠体内的口服吸收比小鼠差,在药物吸收方面大鼠对于药物的筛选更为严格。
图3. 大鼠和小鼠fa × fg数据的对比
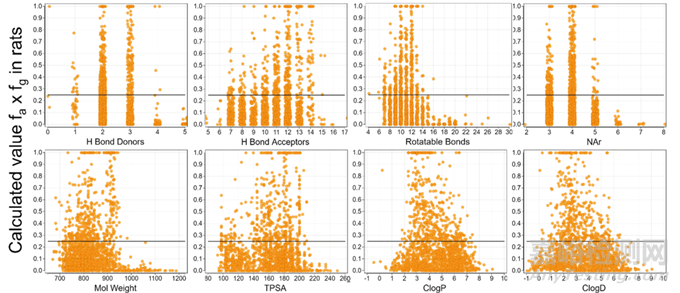
接着研究人员对数据库中PROTAC分子(n=1573)的理化性质与大鼠fa × fg值的关系进行了一个统计(图4),结果概述如下:
氢键供体(HBD)有一个明显的阈值,4个HBD达到fa × fg= 0.25 的上限。在经过对结构的分析后发现,几乎所有(97%)HBD3或所有的HBD4都包含了1-2个分子内被结合的氢键供体。因此,HBD的阈值可以表述为未结合的HBD数量小于等于2个。也就是说当超过2个HBD时,可以引入分子内氢键受体与氢键供体结合,减小HBD的数量限制。
氢键受体(HBA)的上限为15。
可旋转氢键(RB)的上限为14,拓扑分子极性表面积(TPSA)的上限为200 Å2。而之前认为要达到比较好的大鼠口服生物利用度,RB需要小于10,TPSA 要小于140 Å2。PROTAC的数据分析,扩展了原先的范畴。
在分子量(MW)、ClogP/D方面,得出的值也大大超出了原先的类药五原则:MW上限为950;ClogP为1-7;ClogD为0-6。
芳香环数(NAr)的上限为5,上限为4更佳。因为仅有9%NAr为5的化合物得到较好的肠道吸收。
AB-MPS值(AbbVie multiparametric score)的计算方法为AB-MPS= ClogD + NAr + RB,AB-MPS被认为比单个的参数更适合预测低口服吸收的风险,分析得出AB-MPS的上限为20(图5)。
图4. PROTAC分子的理化性质与大鼠吸收的fa × fg值的关系
图5. AB-MPS与大鼠吸收的fa × fg值的关系
表2总结了类药五原则(Ro5),超越五原则(bRo5)以及口服PROTAC分子的理化性质上限,这对于后续的研究具有较大参考意义。从结果上看,与传统的Ro5相比,口服PROTAC分子的HBA、RB、TPSA、MW和ClogP都有了明显的扩展;而口服PROTAC分子的MW、HBA和ClogP与bRo5的上限比较接近。由于PROTAC分子的变色龙性质,对于ClogP和TPSA的限制并没有那么大。相比之下,如果想要达到比较好的吸收,口服PROTAC分子对HBD的限制比较严格。
表2. Ro5 、bRo5和口服PROTAC分子的理化性质上限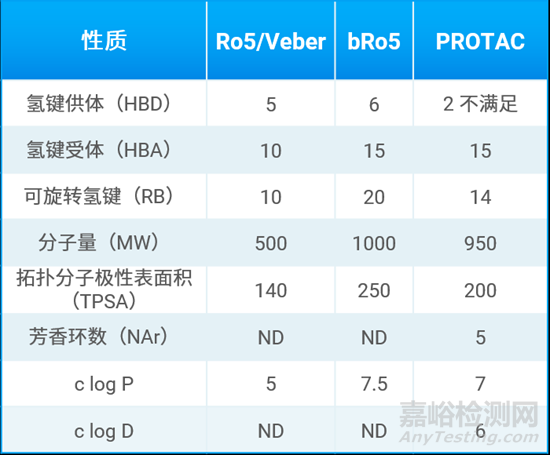
应用口服PROTAC理化性质上限数据指导PROTAC分子设计
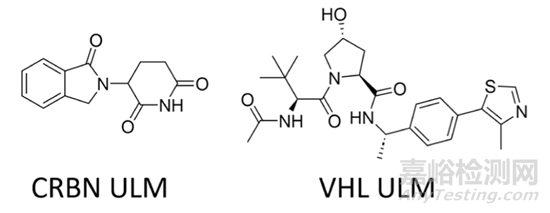
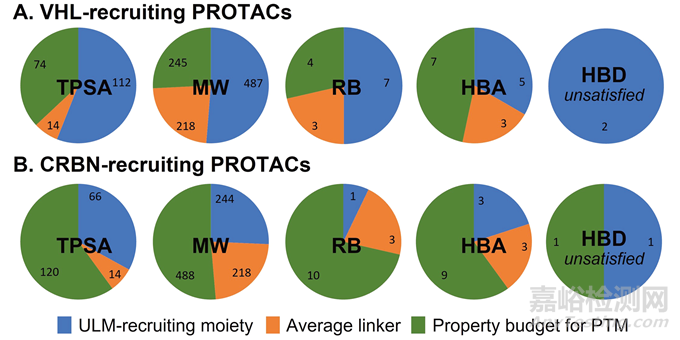
那么在实际研究中该如何应用这些数据呢?首先,研究人员计算了两个代表性的泛素连接酶配体(ULM)的理化参数,即CRBN和VHL(图6),并且计算了20个常用的连接子的平均理化参数,从而建立了一个适合口服吸收的PROTAC分子的靶向蛋白配体PTM的预算图(图7)。在去除了ULM(蓝色部分)和连接子(橙色部分)之后,剩余的即是给PTM留下的空间(绿色部分)。
图6. 两个代表性的ULM的结构图
图7. 基于靶向(A) VHL或(B) CRBN作为E3连接酶配体的口服PROTACs的理化性质预算分析
基于以上分析,可以得到一些结论:
首先,只有明智地选择相对紧凑的ULM,才能得到较好的口服吸收。在口服药物设计过程中避免使用特别大的ULM,比如VHL配体。在数据分析中也可以发现,基于VHL的PROTAC无论是在大鼠还是小鼠中fa × fg都很难达到0.25以上。
其次,即使有一个比较合适的ULM,在PTM的选择上仍然有比较大的限制,比如很难找到一个同时满足含有未结合的氢键供体≤1个、氢键受体≤9个、TPSA≤120 Å2、可旋转氢键数量≤10且分子量≤488的PTM先导化合物。
上述分析中,假定连接子部分经历过优化并具备药物的性质,若后续使用未经修饰的PEG-或烷基型连接子,PTM的可用范围将会进一步缩小。
小结及展望
本文研究发现PROTAC在大鼠体内的口服吸收比小鼠差。而在小鼠之外的种属中,PROTAC的药代数据报道有限。目前也有一些文献报道PROTAC分子在小鼠中有着比较好的口服吸收,包括基于VHL设计的分子。对待这些数据需要十分谨慎,为了支持最终的临床开发,获得跨物种的药代动力学数据是重要的。
本研究采用了一个经验性的阈值,即fa × fg=0.25,但是一个药效足够好的PROTAC,在fa × fg小于0.25时也可能具备了足够的临床暴露量来驱动疗效。当然,这些化合物在临床前和临床开发中可能面临着一些其他的挑战,比如要求在临床前毒理学研究中达到的暴露量数倍于有效暴露量。
在未来,如果需要更好地发挥PROTAC分子的潜力,在分子设计上也提出了几个需求,比如需要更多的筛选工作来找到新的紧凑的ULM和PTM。PTM筛选库中化合物的理化性质值得重新去思考并利用。采用PROTAC 体外ADME实验更好预测体内药代动力学性质,是一项持续的需求。当然也要更多地去关注PROTAC分子在非经肠给药方面的潜力,虽然在早期临床阶段的开发中更倾向于口服给药,但是在某些情况下,出于靶点、患者群体的需求等考量,静脉注射或皮下给药可能更加合适。未来,也期待更多口服PROTAC分子的成功。
参考文献:
[1]Keith R. Hornberger and Erika M. V. Araujo, Journal of Medicinal Chemistry, 2023 66 (12), 8281-8287