前 言
欧盟为了能够更有效的执行MDR法规下规定的警戒的要求,在2023年2月14医疗器械协调小组MDCG发布了“MDCG 2023-3关于医疗器械法规MDR(EU) 2017/745中警戒术语和概念的问题和回答概述”协调文件,该文件为了澄清关于医疗器械法规MDR(EU) 2017/745第 VII 章上市后监管、警戒和市场监管第2节警戒中概述的重要术语和概念。该文件中提出的一些定义从MEDDEV 2.12-1 rev 8医疗器械警戒系统指南中重新引入,并在相关情况下进行了修改,以便与MDR保持一致。
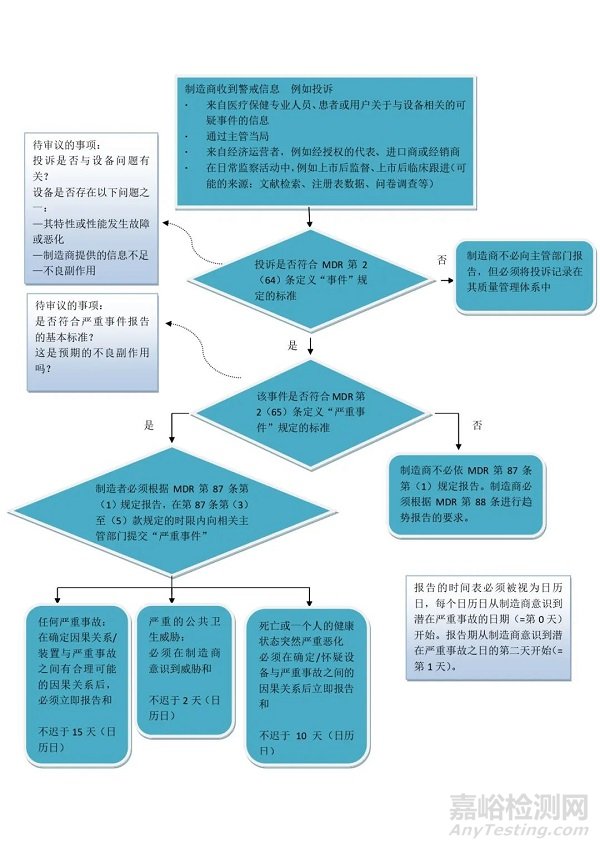
MDR法规下的警戒系统制造商该如何向主管部门报告事件呢,该文件引入了MDR法规下事件和严重事件管理和报告遵循的流程,以更好的来指导制造商如何来管理事件和严重事件。
MDR法规要求
事件
是指市场上可获得的器械特性或性能的任何故障或劣化事件,包括由于人机工程学特征、制造商提供的信息中的任何不足以及任何不期望的副作用而造成的使用错误。
根据MDR要求,事件不向主管当局报告。但是,事件必须在制造商的质量管理体系中记录和考虑,并根据趋势报告的要求报告。
严重事件
是指直接或间接导致、有可能导致或可能会导致以下任一状况的任何事件:
(a) 患者、使用者或其他人员死亡;
(b) 患者、使用者或其他人员健康状态的暂时性或永久性严重恶化;
(c) 严重公众健康威胁。
具体地说,严重事件是直接或间接导致、可能导致或可能导致患者、使用者或其他人死亡或健康状况暂时或永久严重恶化或构成严重公共健康威胁的事件的子集。
报告要求
在欧盟市场上提供器械的器械制造商,应向相关主管机构报告以下内容:
(a)任何涉及在欧盟市场上销售器械的严重事件,但不包括产品信息中清楚地记录并在技术文件中量化的预期副作用外,并根据趋势报告要求报告;
(b)任何有关欧盟市场上销售器械的现场安全纠正措施,若现场安全纠正措施的原因并不仅限于在第三类国家销售的器械,则包括第三国对在欧盟市场上合法提供的器械所采取的任何现场安全纠正措施。
制造商应在制造商与其器械建立了事件之间因果关系后或者发现这种因果关系合理时,立即报告任何严重事件,这一时限不迟于其意识到严重事件后的 15 天。
若出现严重的公共卫生事件,则应立即报告,且不迟于制造商察觉到此威胁后 2 天。
若出现人员死亡或健康状况意外严重恶化,该报告应在制造商确认或可疑器械与严重事件之间的因果关系后立即提供,且不迟于制造商察觉到该严重事件之日后 10 天。
事件和严重事件管理流程