您当前的位置:检测资讯 > 行业研究
嘉峪检测网 2022-05-11 15:59
摘要 目的:在连续制造技术越来越多地应用在药品领域这一背景下,综述相关法规指南从概念探索、正式发起至发布的发展历程,介绍全球多个采用连续制造技术生产药品获批上市的概况,探讨促进我国业界和监管机构借鉴全球药品连续制造发展经验。方法:通过对比传统的批量制造技术以分析连续制造具有的优势和面临的挑战,结合对全球监管指南制定过程中各监管机构对策的梳理,研究相关共识的发展考量和意义。结果:药品连续制造监管发展已经进入了新的时代,我国相关法规指南的制定和产业技术水平提升需要借鉴全球发展的经验,特别是国际人用药品注册技术协调会及美、欧、日等国家药品监督管理机构或国际组织的现有指南在批定义、工艺验证、稳定性等方面的监管对策,为该新兴技术的监管科学研究提供理论基础。结论:通过对药品连续制造全球监管发展现状的综述和思考,为我国相关法规、技术指南和标准的制定提供参考,并希望为产业发展发挥促进作用。
1药品连续制造概述
药品连续制造(Continuous Manufacturing,CM)是新时代制药工业智能制造重要的先进发展方向之一,理论上既可以应用于原料/原液,也可以应用于制剂。从该技术自身特点来看,其定义一般是指将原料连续地输入和转换,而加工后的输出材料连续地从系统中移出的过程[1]。其中,“系统”是指由2个或更多单元操作组成的集成[2]。连续制造技术已在汽车、食品、消费品和石化行业成功应用多年,以提高制造效率和降低成本;而相比之下,在制药领域则处于起步阶段,传统批量制造技术仍是制药行业生产的主流模式[3]。随着2021年7月国际人用药品注册技术协调会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)发布了Q13指南草案[4],开始向各监管成员机构征求意见,药品连续制造监管发展已经进入了新的时代。
Q13指南所指“连续制造”涉及生产过程中输入物料的持续投料,物料的持续转化,以及输出物料的同步去除。仅单个单元操作为连续,且该单元变更对下游和上游不产生直接且即时影响的情况,通常不看做真正意义的“连续”。监管概念方面,连续制造的模式可分3类,即“批量+连续”“原料连续+制剂连续”以及“端到端连续”。虽然也需要明确批次,但不同于批量制造的是,连续制造可以通过输入/输出物料、生产时间等因素来定义批次。此外,指南要求考虑整体方法制定控制策略,科学地研究生产输出的变更、连续工艺确证等新的方法,鼓励充分运用过程分析技术(Process Analytical Technology,PAT)和实时放行检验(Real-Time Release Testing,RTRT)等。对于指南涉及的定义和监管概念、科学方法在本文第3节进行了更具体的分析。
连续制造技术应用于药品的优势与挑战并存。相对于传统批量制造技术,连续制造的优势如表1所列[1,3,5-9]。

连续制造技术从药品研发探索到真正的引起业界广泛关注经历了十余年的时间,尽管该技术具有显而易见的优势,并得到监管机构的持续鼓励,但在制药行业的应用仍偏于平缓,其潜在的挑战可想而知。在明确的监管指南发布以前,开展连续制造工艺验证并申请注册获得上市批准的投入可能超过了获益,在多地区注册时这种困境更加突出[10]。见表2[6,8,11-13]:

无论如何,新技术的应用是行业发展的大趋势。2015年至2021年,美国、欧盟以及日本陆续批准了涉及3类工艺生产线、4家公司的9个口服固体制剂上市[1-14]。总体上,连续制造在各类药品生产中的应用均呈现增长趋势,全球多地区建设了相关的生产设施。近年来,该领域科学技术快速发展促进全球业界实践以及监管经验的积累,对我国药品连续制造相关法规指南的制定具有重要借鉴意义[15]。
2全球监管发展概况
2.1 ICH
ICH较早前即倡导秉持QbD原则的连续制造技术,已发布的系列指南如Q8、Q9、Q10、Q11以及Quality-IWG,尽管没有直接提及连续制造,但已为制药行业QbD的范围和定义提出了要求[1]。而连续制造工艺的开发过程基于对产品和工艺过程更深入的理解,并且需要更为先进的生产控制手段以提升药品质量,因此,ICH认为连续制造是QbD理念的完整体现[16]。2018年ICH开始逐步推进连续制造指南制定工作,直至目前,已有11家监管机构和行业组织参加了该工作组,包括国际药品认证合作组织(Pharmaceutical Inspection Convention and Pharmaceutical Inspection Co-operation Scheme,PIC/S)。通过专家工作组一系列的研究讨论(见图1)[17-20],2021年7月27日,ICH签署了Q13(Continuous Manufacturing of Drug Substances and Drug Products)指南第2阶段文件,并由ICH监管成员发布,以征求公众意见。这对于药品连续制造的发展具有里程碑意义。
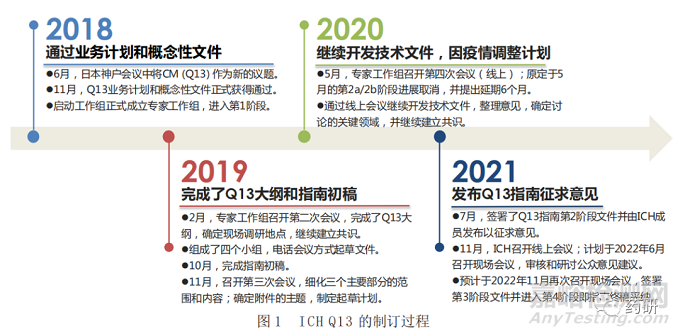
ICH 预期,Q13 指南通过协调采用连续制造生产的原料药和制剂生产的监管期望,增加相关活动在全球实施的可行性 [21]。2021 年 10 月 18 日,我国国家药品监督管理局药品审评中心官网也发布了关于公开征求 ICH 指导原则《Q13:原料药和制剂的连续制造》意见的通知。
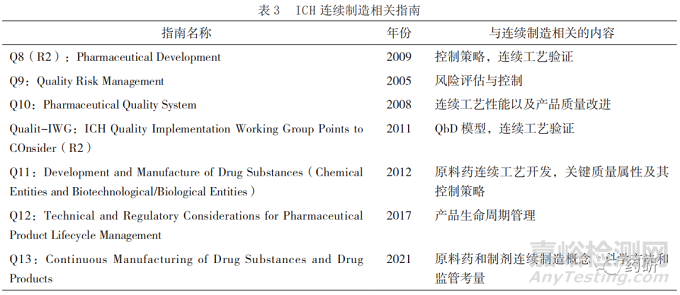
ICH 发布的与药品连续制造相关的指南见表3[22-27]。
2.2 美国食品药品监督管理局
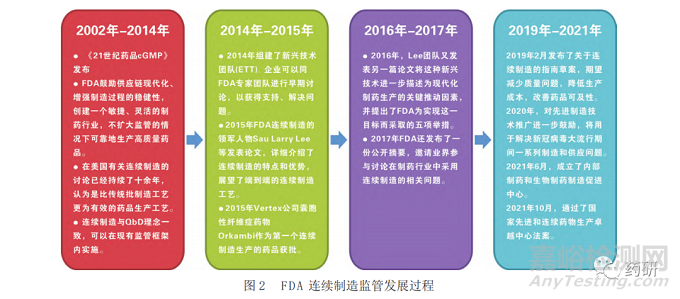
自2002年《21世纪药品cGMP》发布以来,美国食品药品监督管理局(U.S. Food and Drug Administration,FDA)一直鼓励行业采取措施对药品生产进行持续改进[28],其目标是使供应链现代化、增强制造过程的稳健性,创建一个敏捷、灵活的制药工业体系,在不扩大监管水平的情况下仍能可靠地生产高质量的药物[6]。FDA认为连续制造与上述目标非常契合,符合QbD理念,并且可以在现有的监管框架内实施[15]。美国连续制造的监管经历了十余年的发展,积累了一定的经验(见图2)[2, 5, 29-31]。FDA发布的与药品连续制造相关的相关指南见表4[2,32-35]。

2.3 欧洲药品管理局、英国药品和保健品管理局
欧洲药品管理局(European Medicines Agency,EMA)在 2017 年一次公开会议上指出 [36],监管机构支持药品创新制造技术。虽然 EMA 暂没有提供具体指南,但目前的监管框架足以支持连续制造。EMA 已经通过集中审批程序审评了 2 个连续制造应用,其中一个是 EMA-FDA QbD 试点。EMA 已成立 2 个小组,为推进药品创新技术提供支持,一个是 PAT 小组 [37],一个是创新专责小组 [38]。后者涵盖了新兴疗法和技术,汇集了质量、安全性、有效性、药物警戒、科学建议、孤儿药和良好实践合规以及法律和监管事务方面的专家。与其他监管机构的类似组织一样,EMA 的这些小组在连续制造发展过程中鼓励早期对话。
英国药品和保健品管理局(Medicines and Healthcare products Regulatory Agency,MHRA) 也在积极推动包括连续制造在内的制药创新技术。作为 PIC/S 的成员,MHRA 通过“准入联盟”与澳大利亚、加拿大、新加坡和瑞士的监管机构建立联系,共同促进更大的监管协作和监管要求的一致性,MHRA 创新办公室 [39] 是其联络点,而制造过程的创新是该组织最感兴趣的领域之一。
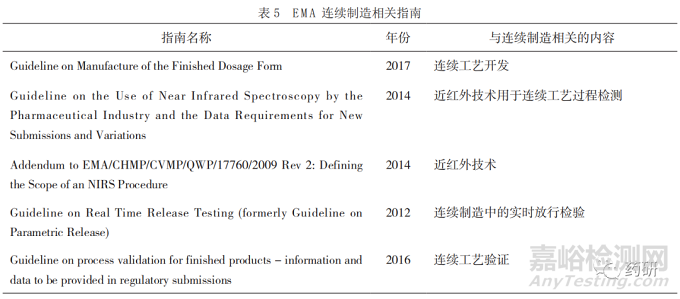
EMA 发布的与药品连续制造相关的相关指南见表 5[40-44]。
2.4 日本独立行政法人医药品医疗器械综合机构
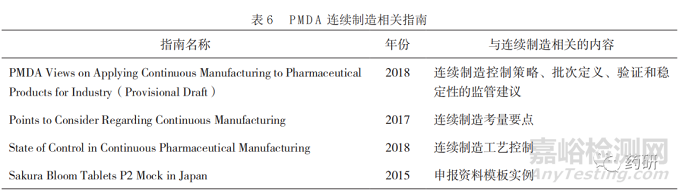
日本独立行政法人医药品医疗器械综合机构(Pharmaceuticals and Medical Devices Agency,PMDA)与欧美监管机构一样高度支持连续制造 [45]。2016年,PMDA 成立了创新制造技术工作组(Innovative Manufacturing Technology Working Group,IMT-WG),以促进创新制造技术的引进,并于 2018 年批准了一个连续制造片剂产品上市申请。PMDA 对于将该技术应用于药品生产的相关考量包括对控制策略、批次定义、验证和稳定性实验等多个方面 [46-57]。
PMDA 发布的与药品连续制造相关的相关指南见表 6[47-50]。
2.5 其他机构或组织
2.5.1 美国药典委员会
美国药典委员会(United States Pharmacopeia,USP)一直积极与学术研究中心、原研药企、仿制药企业和监管机构合作,以推动药品连续制造标准化工作,并开发合作平台。2016 年,USP 成立药品连续制造专家小组,持续与药品连续制造利益相关方和领军人物进行接洽 [51]。
USP 在标准化成分表征方法、规范产品表征方法、规范设备性能要求、标准化传感方法、标准化控制能力、规范产品 / 工艺开发方法方面开展了一系列的讨论,并持续组织相关培训 [52]。2018 年,发布了“美国药典委员会对药品连续制造的考量”,就行业关注的多方面问题进行了讨论和回顾,USP专家小组认为该文章是第一次尝试将药品连续制造领域使用的大量术语和定义进行标准化 [53]。2021年,提出了促进美国采用 CM 的政策概念,建议制定政策以建立一个更具弹性的供应链,即使在疫情或其他危机时期也可以帮助确保为患者持续提供安全、高质量的药品 [54]。
2.5.2 美国材料与试验协会
2014年, 美国材料与试验协会(American Society for Testing and Materials,ASTM)发布了“制药工业连续工艺应用的标准指南”,包含了连续制造元素的较全面信息,是关于药品连续生产的较早的、简明且相关性较高的指南 [55]。
2.5.3 国际制药工程协会
国际制药工程协会(International Society for Pharmaceutical Engineering,ISPE)口服固体实践委员会连续制造分委会正在计划起草相关的良好实践指南,总结团队在过去几年里开发的成果,以建立设备需求、确定协调和灵活集成的可行性,并对改进现有设备的具体方向提出建议,以便与未来的连续制造平台协同工作 [56]。
ISPE 在一篇报道 [57] 中指出,通过对部分企业的药品连续制造申报上市情况调研得出结论,监管机构是支持创新的,药品连续制造生产的产品在全球的申报没有障碍;行业经验是尽早与监管当局开展高频次的沟通;邀请监管机构实地调研连续制造生产现场,对相互了解申报审评策略是有益的;监管机构鼓励早期参与专门的监管互动渠道;此外,美国正在加快变革的速度。
3各监管机构关于连续制造关键点监管考量横向对比
连续制造监管考量具有一定的地区特点,笔者对全球监管机构指南中的一些关键点进行了分析。在批与批量描述、控制策略、工艺验证、稳定性、批工艺到连续制造的变更、申报资料要求等几个方面,横向对比显示,各监管机构关于连续制造关键点监管考量各有侧重(见表 7)。考虑到欧盟没有专门的连续制造指南,横向对比的内容以 ICH、FDA、PMDA 的连续制造指南为主。





从表 7 可以看出,相对来说,ICH Q13的原则性更宽泛、包容度更高,FDA、PMDA 的要求则更为具体,对相关研究的深度和细节要求更高。在申报资料指导方面,ICH Q13 的发布时间更晚,相对更详细。
4连续制造监管经验的思考
纵观全球药品连续制造监管发展历程,笔者认为有很多方面的经验值得我国参考借鉴。以下将从指南制订策略、监管提前介入、创新知识储备、国家标准制订、行业协会促进、委托研制、仿制药的应用几个方面进行讨论。
4.1 指南制订策略
ICH Q13 指南的发布有助于协调我国和其他国家地区的监管期望以及审评与监管尺度的一致性。专家工作组在 Q13 指南制定过程中发挥了关键作用,纳入在连续制造的技术和监管方面具有足够背景、专业知识和 / 或经验,并具有创新思维的监管和行业代表,以提升工作基础水平和可行性。Q13的制订与成员国审评审批同步开展,过程中尽可能形成行业和监管共识,以框架性的“正文”加上细分领域“附录”的形式兼顾包容性和具体性,还充分借鉴药品以外其他行业(例如石化、食品、烟草等)基础科学方法和连续制造知识。建议我国借鉴全球监管机构的指南制订策略,采用专项小组、合作课题研究等方式,扩充专家工作组的专业领域和人才力量,结合我国国情和实际申报品种审评进度,包容审慎,逐步推进细分领域指导原则的制定。
4.2 监管提前介入
美国、欧洲和日本对业界开展新兴技术研发均加以鼓励,认可连续制造可以在现有的监管框架内实施。在化药、生物制品技术审评,以及监管检查机构分别建立新兴技术团队,与业界加强早期沟通。
以 FDA 为例,截至 2021 年已接受上百个涉及广泛创新技术的申报、组织了上百个研讨会,在其“新兴技术计划”(ETP)的工作量已经增加到接近饱和情况下,及时升级 ETP 2.0 来满足提前介入需求。我国也有监管早期沟通的渠道,建议进一步借鉴国外经验增强新兴技术团队力量,明确权责和工作制度,并加大提前介入力度。
4.3 创新知识储备
在新冠疫情期间,为解决药品制造和供应链问题,FDA 等机构对先进的制造技术推广给予进一步鼓励,并通过立法的方式在其国内科研机构建立国家连续生产卓越中心等创新技术研发项目,促进制药行业的先进生产。对未来即将出现的新技术发展方向较早进行预判,并积极储备知识和经验,值得我们借鉴。
4.4 国家标准制订
USP 等机构积极与业界合作,开展对连续制造输入和输出物料 PAT、RTRT 等方面标准开发和制定。我国国家药典委员会在药品国家标准制定方面发挥了核心作用,《中华人民共和国药典》在批次定义、方法学验证、稳定性考察方面都有相应的规范或指导原则,对于我国药品连续制造的发展方向有较高的影响力,参考国外的相关经验有助于进一步优化相关国家标准的制修订,不断完善我国在这一领域的监管框架。
4.5 行业协会促进
ASTM、ISPE 等在前沿技术领域帮助其会员企业和监管机构搭建沟通桥梁,极大地提高了行业与监管共识形成的效率。我国的行业协会对制药工业界的发展也起到了重要的协调和促进作用,在连续制造监管科学研究和行业共识方面也应当充分发挥行业协会的优势。
4.6 合同研制
全球多数连续制造项目由大型制药公司与科研机构合作发起,相关主管部门对科研成果成功转化的鼓励发挥了重要作用。合同开发和制造或 组 织(Contract Development and Manufacturing Organisations,CDMO)积极参与药品连续制造研发,并与学术机构商讨共同成立研究中心,这将加快成果转化效率,有助于形成可复制推广的新工艺模型 [58]。更多的受托研发和制造力量加入产品生命周期生态圈,可以让连续制造业界更具活力和纵深。我国药品上市许可持有人制度已有一定基础,建议进一步鼓励相关优质研发生产服务机构提高对连续制造技术的供应支持。
4.7 仿制药的应用
从全球监管发展过程来看,连续制造更多的应用于新药、新车间,或产能提升需求迫切的已上市产品。对于仿制药企业,其对连续制造的兴趣点不同于原研药企,可能因初始投入大、技术要求高而参与意愿有限 [59],全球仿制药连续制造参与度明显滞后于原研药。我国仿制药体量较大,在群众用药可及性方面发挥了巨大的作用,但因利润空间缩小等原因,企业难以充分增加研发投入,如何提升仿制药企业的连续制造创新积极性以进一步提升质量降低成本,促进更广大患者受益,需要深入思考,建议对仿制药生产技术创新加以政策鼓励,并加大科学技术指导支持的力度。
5展望
随着 ICH Q13 的发布,加之欧美日监管机构已有的指南,药品连续制造技术的引入已经有了充分的法规支撑。但在一些具体方面,还需要更多的细则出台。通过对各监管机构发布的指南等文件的分析,并与 ICH Q13 进行对比发现,各监管机构在连续制造的监管考量方面具有一定的地区特点,在要求细节方面也有差异,体现了各自的侧重点以及相应地区产业发展的特色,同时也在监管策略方面提供了很多的宝贵经验,这是我国开发相关指南可以参考借鉴的。
ICH Q13 总体还是相对框架性,在各国具体的实施过程当中,监管机构对相关产业的鼓励、引导,广泛深入的互动和沟通,对药品连续制造取得成功起到了关键作用,这也启示我们,制药技术改革创新不但需要监管机构持续重视,更需要社会各界的共同参与。尽管还在不断完善中,监管指南的发展已经在对药品连续制造技术的规范发挥着重要推动作用,而且可以预见,药品连续制造将迎来更广泛、更深入的应用。对全球相关领域进展情况进行深入调研,本着鼓励创新、包容审慎的原则制定药品连续制造监管规则,对推进我国新兴产业发展和监管策略的完善具有现实意义。

来源:Internet


