您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-04-28 14:29
摘要:目的 建立非布司他片的放大制备工艺,并评价自研制剂与原研制剂的体外一致性。方法 采用流化床工艺制粒,按2020年版《中国药典(四部)》溶出度和释放度测定法第二法(桨法)测定溶出度,以相似因子(f2)法评价自研制剂与原研制剂的相似性。结果 所制备的非布司他片f2为76~91,均大于50。结论 流化床制粒放大工艺可行,自研制剂与原研制剂的体外溶出行为一致。
非布司他为黄嘌呤氧化酶抑制剂,临床用于治疗高尿酸血症伴痛风,在降低血尿酸的同时还能改善血脂代谢异常,安全性较好[1-2]。非布司他片由日本武田北美制药公司研发,最早上市于美国[3]。体内外相关性研究可预测仿制制剂与原研制剂相似的体内药代动力学行为,降低研发成本,缩短研发周期[4-5]。本研究中开发了非布司他片的放大制备工艺,并依据原国家食品药品监督管理总局发布的相关指导原则[6-7],开展了与原研制剂的体外一致性评价。现报道如下。
1仪器与试药
1. 1 仪器
708-DS型全自动溶出仪(美国安捷伦公司);Waterse2695型高效液相色谱仪(美国Waters公司),配有Waterse2695 型四元梯度泵、Waters 2489 型紫外检测器、自动进样器;MSA225s-100-DA型分析天平(德国Sartrious公司,精度为0. 01mg);WBF-60 型多功能流化床(重庆英格造粒包衣技术有限公司);ZLZ450型真空整粒机(武汉恒达昌机械设备有限公司);HF300 型方锥混合机、GZPL40C型高速压片机(南京邦斯特制药设备有限公司);BGB-75C 型包衣机(宜春万申制药机械有限公司)。
1. 2 试药
非布司他对照品(批号为101278 - 202003,纯度为100. 0%),十二烷基硫酸钠(分析纯,批号为20197656),均购自中国食品药品检定研究院;非布司他片(商品名Feburic < 菲布力 > ,安斯泰来制药 < 中国 > 有限公司,批号为8155,规格为每片40mg);非布司他自研制剂(批号分别为 2012404,2012405,2012406,规格为每片40mg);非布司他原料药[国内某制药厂,晶型A,粒 径(D90)<50µm,批号为20198234];乳糖(200目,德国美剂乐公司,批号为6397269);PC-10型预胶化淀粉(日本旭化成株式会社,批号为5RJ071);羟丙基纤维素(日本曹达株式会社,批号为NCD-2993);SD - 711型交联羧甲基纤维素钠(DuPont Nutrition USA Inc. ,批号为 3201092c34);硬脂酸镁(曲阜市天利药用辅料有限公司,批号为20182324);胃溶型薄膜包衣预混剂(上海卡乐康包衣技术有限公司,批号 02F690004-CN);乙腈(色谱纯,德国Merck公司,批号为HBN0911);冰醋酸(分析纯 ,国药集团化学试剂有限公司 ,批号为1000208);水为纯化水。
2方法与结果
2. 1 非布司他片放大制备工艺
原辅料过筛:将非布司他微粉化,测得合适的粒径范围,其他辅料过40目筛,按处方量称取,备用。
物料混合:配制6%羟丙基纤维素溶液,依次将乳糖、非布司他、预胶化淀粉加入混合机中混合30min,得预混料。
流化床制粒:将预混料加入流化床,参数设置为,进风温度(80 ± 10)℃,蠕动泵转速80~120 r / min,风量750~1200 m3 / h。制粒完毕后,待干燥失重不超过2. 0%,停止干燥。干燥结束后继续抖袋60s,收料完毕后,采用固定真空整粒机整粒,筛网孔径为2. 0 mm。
总混:外加辅料交联羧甲基纤维素钠,混合10min;加入硬脂酸镁,混合5min。
压片:采用直径为9. 0mm 的圆形双弧浅凹冲对总混颗粒压片。控制片芯硬度为40~70N。
包衣:称取处方量胃溶型薄膜包衣预混剂适量,缓慢加入纯化水中,搅拌均匀(≥ 45 min),配制12%的包衣液。打开包衣机,包衣前将素片放入包衣锅内,调整喷枪角度,设置主机转速为2r / min,预热磨片1~3min,取100片素片,称定质量,设置参数为,进风温度(60 ± 10)℃,主机转速2~5r / min,蠕动泵转速30~600r / min,开枪压力0. 20~0. 40MPa,雾化压力0. 20~0. 40MPa,开始包衣,包衣增重约3%,停止喷浆,将主机转速调至2 r / min,干燥15~25min,停止加热,待冷却至出风温度不超过35 ℃,出料,即得非布司他片包衣片。
2. 2 溶出介质确定
测定非布司他在不同pH(pH = 1. 0,2. 0,3. 0,4. 0, 5. 0,5. 2,5. 4,5. 6,5. 8,6. 0,6. 2,6. 4,6. 6,6. 8,7. 0, 8. 0)条件下的溶解度,结果非布司他为 pH - 溶解度依赖药物,几乎不溶于水和酸性溶液,溶解度随pH的增大而逐渐增大。为建立具备区分能力的溶出曲线,测定非布司他在水及0. 1%,0. 3%,0. 4%,0. 5%,0. 6% 十二烷基硫酸钠溶液中的溶解度,各pH条件平行测定3次,结果非布司他在 0. 5% 十二烷基硫酸钠溶液中能达到漏槽条件。故最终确定溶出介质为0. 5%十二烷基硫酸钠溶液。
2. 3 含量测定
2. 3. 1 色谱条件
色谱柱:ACE Excel 3 C18 - AR柱(50 mm × 4. 6 mm, 5 µm);流动相:乙腈 - pH 3. 0醋酸溶液(50∶50,V / V);流速:1. 0 mL / min;检测波长:317 nm;柱温:40 ℃;进样量:20 µL。
2. 3. 2 溶液制备
取非布司他对照品适量,精密称定,加乙腈 - 水溶液(60∶40,V / V)溶解,稀释成每1mL含非布司他40µg的对照品溶液。取样品,加0. 5% 十二烷基硫酸钠溶液900mL,按 2020年版《中国药典(四部)》0931溶出度与释放度测定法第二法(桨法)测定,设置转速为50r / min,分别取溶出 10,15,20,30,45,60 min时的溶液适量,即时补充相同体积 0. 5% 十二烷基硫酸钠溶液,滤过,取续滤液,即得供试品溶液。按处方量称取不含主药的辅料,混匀,取细粉300mg,加0. 5% 十二烷基硫酸钠溶液900 mL,超声(功率为600W,频率为40kHz)10min,滤过,即得空白辅料溶液。
2. 3. 3 方法学考察
专属性试验 :分别吸取2. 3. 2项 下3种溶液及0. 5%十二烷基硫酸钠溶液、乙腈 - 水溶液(60∶40,V / V),按2. 3. 1项下色谱条件进样测定。结果供试品溶液色谱中,在与对照品溶液色谱相同位置处有相应色谱峰,其他溶剂均无干扰,表明方法专属性良好。色谱图见图1。
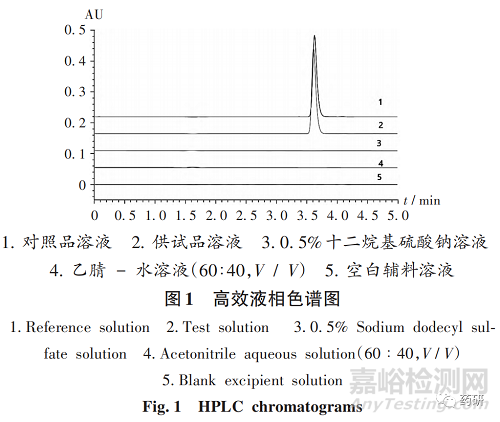
线性关系考察:取非布司他对照品适量,精密称定,加乙腈 - 水溶液(60∶40,V / V)溶解,稀释成每1mL约含非布司他0. 1 mg 的线性试验贮备液。精密量取线性试验贮备液1,2,5,8,10,15 mL,置100 mL容量瓶中,加乙腈 - 水溶液(60∶40,V / V)稀释至刻度,摇匀,得10%,20%,50%,80%,100%,150% 系列溶液。精密量取上述溶液各20µL,按2. 3. 1项下色谱条件进样测定,记录色谱图,以质量浓度(X,µg / mL)为横坐标、峰面积(Y)为纵坐标进行线性回归,得回归方程Y = 1 577 459. 94X +38 998. 32(r = 0. 999 9,n = 6)。结果表明,非布司他质量浓度在0. 99~14. 85 µg / mL范围内与峰面积线性关系良好。
定量限与检测限确定:取线性关系考察项下线性试验贮备液适量,逐级稀释,按 2. 3. 1 项下色谱条件进样测定6次,分别以信噪比(S / N)的10倍和3倍 为定量限和检测限。结果定量限和检测限分别为2. 59ng / mL和0. 78ng /mL。
日间精密度试验:取2. 3. 2项下溶出60min时的供试品溶液适量,由不同分析人员在不同时间采用不同溶出仪,按2. 3. 1项下色谱条件进样测定,记录峰面积,并计算溶出度。结果平均溶出量为97. 73%,RSD为0. 93% (n = 6)。
稳定性试验:取供试品溶液适量,分别于室温下放置 2,4,6,8,12,24 h 时按2. 3. 1 项下色谱条件进样测定,记录峰面积。结果的RSD小于1. 00%(n = 6),且未发现其他新的未知峰,表明供试品溶液在室温下放置24h稳定性良好。
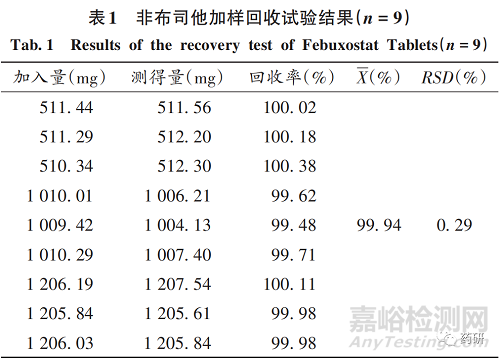
加样回收试验:取非布司他对照品20,40,48 mg,精密称定,分别置200 mL容量瓶中,按处方比例加入空白辅料230mg,加乙腈 - 水溶液(60∶40,V / V)适量, 超声使溶解,稀释至刻度,摇匀,滤过,精密量取续滤液5mL,置100mL容量瓶中,用流动相稀释至刻度,摇匀,作为低、中、高3个浓度水平的加样回收试验溶液,每个浓度水平平行制备3份。按 2. 3. 1项下色谱条件进样测定,结果见表1。
2. 4 体外一致性评价
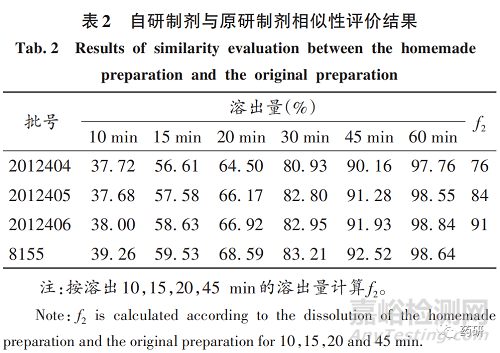
取3批自研制剂与1批原研制剂,各12片,溶出介质为0. 5% 十二烷基硫酸钠溶液,按2020年版《中国药典(四部)》0931溶出度与释放度测定法第二法(桨法)测定,比较溶出曲线。采用相似因子(f2)法评价相似性,结果见表2。可见,3批自研制剂的f2最低为76,均大于50,表明相似性较好。
3讨论
3. 1 流化床制粒
流化床制粒可在一台设备内一步完成混合、制粒、干燥、包衣,具有简便、机械化程度高的特点。处方中选择的辅料适合流化床的工艺特点,混合工序能保证颗粒的均匀性,表明本工艺具有良好的可行性,适用于放大生产。物料温度是制粒的关键步骤,温度过高,雾化后的液滴来不及黏附在粉末上,即被干燥成细粉和细颗粒;温度过低,粉末表面的浸膏来不及干燥,甚至形成团块,出现“塌床”现象。本研究中进风温度为(80 ± 10)℃,蠕动泵转速为80~120r / min,可合理控制物料温度。原料药难溶于水,为增加其溶出度,同时拟合原研制剂的溶出行为,将非布司他原料药经微粉化处理,得到≤25µm、≤50µm、≤75µm、≤100µm 4个粒径的原料药,分别制成片剂后考察其溶出行为。结果该溶出方法能区分这4种不同粒径原料药生产的制剂。
3. 2 溶出度测定方法
本研究中采用高效液相色谱法测定溶出度,同紫外光谱法相比,其测定灵敏,结果准确,更能保证f2测定的准确性。方法学考察结果表明,本方法专属、灵敏、准确,符合2020年版《中国药典(四部)》中相关要求[8]。
美国食品药物管理局发布的橙皮书中收载的溶出度方法,以pH6. 0磷酸盐缓冲液为介质,采用桨法,以50r/min的转速进行测定。在该条件下,自研制剂于30min时溶出超过80%,不具备区分能力。采用紫外光谱法, 以0. 5% 十二烷基硫酸钠溶液为溶出介质[9],区分能力仍然不够。非布司他为BCSⅡ类低溶解性-高渗透性药物,溶出速率是药物吸收的限速步骤[10],样品投入溶出杯后迅速崩解成细小颗粒。故确定了2. 2项下的溶出介质,非布司他满足漏槽条件,且与肠道生理条件下的pH相近,能合理模拟体内的溶解环境。
以不同品牌十二烷基硫酸钠为溶剂时,溶出度测定结果存在差异[11],故其品牌不能随意更换,且批号应尽量一致。按本试验条件测定原研制剂的溶出度,结果在45~60min 时溶出量达85%,且曲线上无拐点和突释,可用于区分溶出能力。
3. 3 原料药选择
原料药的晶型和粒度严重影响药品的溶出,故研究原料药的晶型十分必要。经调研,本品具有6种晶型[9],但不同晶型的表观溶解度和饱和溶解度无显著差异,且晶型不影响本品的有效性。故可使用适合工业化生产的晶型A。
3. 4 方法评价
本研究中采用流化床工艺制备非布司他片,其与原研制剂的 f2均大于50,体外溶出曲线相似,适用于非布司他片的制备。

来源:Internet


