您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2025-08-03 23:49
近日,FDA发布了PROGRAM 7356.002F《API 检查指南》,文件为 API 生产工厂提供全面的 CGMP 检查范围,并将于2025年9月2日实施。
文件包含以下内容:
PART I – BACKGROUND
第一部分 背景
1. Introduction
介绍
2. Applicable Statute, Regulations, and Guidances
适用法律、法规和指南
3. Scope of APIs Covered by This Program
本程序涵盖的 API 范围
4. Definitions
定义
PART II –IMPLEMENTATION
第二部分 实施
1. Objective
目的
2. Program Management Instructions
程序管理说明
A. Selecting Sites
场地选择
B. Selecting Investigators
检查员的选择
C. Selecting Inspection Types
检查类型的选择
D. Selecting Systems
被检查系统的选择
E. Selecting APIs
API选择
F. Selecting Profile Classes
配制文件类型选择
PART III –INSPECTIONAL
第三部分 检查
1. Operations
操作
A. Inspection Approaches
检查方法
B. System Coverage
被检查系统的覆盖范围
C. Preparing the Inspection Strategy
准备检查策略
2. Reporting
报告
A. Special Instructions for Responding to a Form FDA 483
回复 FDA 483 的特别说明
PART IV –ANALYTICAL
第四部分 分析
PART V –REGULATORY/ADMINISTRATIVE STRATEGY
第五部分 监管/行政策略
PART VI –REFERENCES, ATTACHMENTS, AND PROGRAM CONTACTS
第六部分 参考文献、附录和程序联系方式
1. References
参考文献
2. Attachments– None
附录 无
3. Program Contacts
程序联系方式
A. CDER
B. OII
文件提及,对API制造商的检查应由经验丰富的检查员进行,他们接受过与API生产工艺类型或API制造商进行的工艺(例如发酵、化学合成)相关的充分教育和培训。应酌情考虑将化学和微生物学专业人员纳入API检查组,特别是评估实验室操作(例如,分析方法评估、 分析数据、实验室程序、仪器)和评估用于建立杂质谱、发酵制造工艺和化学合成的复杂多步骤工艺的分析方法。
文件将GMP检查分为两种类型:监督检查和有因检查。有因检查包括:(1)CGMP合规跟踪检查,以确认采取监管行动后的纠正措施;(2)针对特定事件或信息(例如,现场警报报告、生物制品缺陷报告、行业投诉、召回、其他产品缺陷)进行CGMP检查,这些事件或信息使制造操作、设施、工艺或API的合规性或质量受到质疑。
文件详细介绍了质量体系、设施与设备系统、生产系统、物料系统、包装与标签系统和实验室控制系统的检查内容:
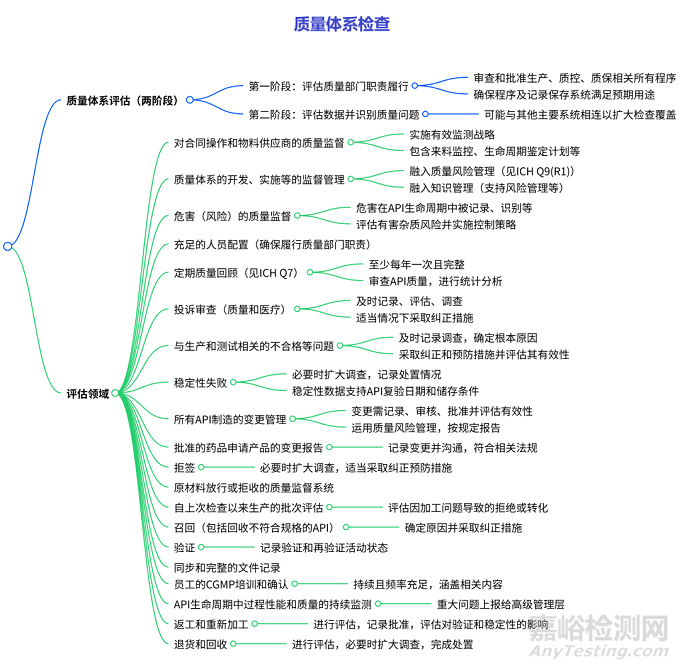
质量系统的检查
分为两个阶段:
第一阶段:调查人员评估质量部门是否履行了审查和批准与生产、质量控制和质量保证相关的所有程序的责任,同时评估质量部门是否确保这些程序及相关记录保存系统足以满足预期用途。
第二阶段:调查人员对数据进行评估,识别任何质量问题,且该阶段可能与其他主要系统相连以扩大检查覆盖范围。
涵盖多个方面,包括对合同操作和物料供应商的质量监督(需实施有效监测战略等);质量体系开发、实施等的监督管理(融入质量风险管理和知识管理);危害(如交叉污染、有害杂质)的质量监督(在 API 生命周期中进行记录、评估等);充足的人员配置;定期质量回顾(至少每年一次,含 API 质量审查和统计分析);投诉审查(及时记录、调查等);生产和测试相关的不合格等问题的处理(及时调查、采取纠正预防措施);稳定性失败的应对(扩大调查、记录处置等);API 制造的变更管理(记录、审核、评估风险等);批准药品的变更报告(符合法规);拒签、原材料放行 / 拒收的质量监督;自上次检查以来生产批次的评估;召回(确定原因、采取纠正措施);验证(记录相关活动状态);同步完整的文件记录;员工 CGMP 培训(持续且涵盖相关内容);API 生命周期中过程性能和质量的监测(重大问题上报);返工和重新加工(评估、记录等);退货和回收(评估、调查等)。同时,文件多次引用 ICH 相关指南及 FDA 法规作为参考依据。
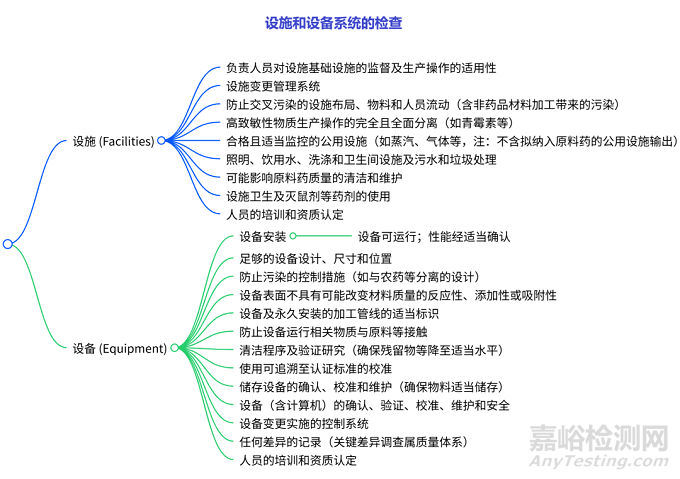
设施和设备系统的检查
< >设施检查:涉及人员监督、变更管理、布局与防污染、高致敏性物质生产隔离、公用设施监控、基础设施(照明、水系统等)、清洁维护、卫生与药剂使用、人员培训等。设备检查:包括安装与性能、设计尺寸位置、防污染控制、表面特性、标识、防物质接触、清洁验证、校准标准、储存设备维护、设备全流程管理(含计算机)、变更控制、差异记录、人员培训等。

物料系统的检查
涵盖人员培训与资质、物料及容器的标识、储存、隔离与先进先出使用,代表性样品采集与检验,关键物料供应商审计监控,不合格物料处理,工艺用水和气体系统适用性,计算机化流程的确认验证与安全,物料处理变更管理,批次分发及差异记录,以及起始物料等的风险管理计划等内容的检查。

生产系统的检查
包含人员培训、生产程序管理、关键活动控制、偏差处理、产量监控、工艺验证、API 安全控制、生产时限、设备标识、规格合理性、过程控制与检测、物料回收、交叉污染防控、自动化流程验证、统计评估、变更管理、批记录管理及有害杂质风险控制等的检查,详细列出了需要评估的内容及相关要求。

包装和标签系统的检查
包含人员培训资质、物料验收、程序制定与执行、变更管理、标签储存与控制、记录管理、成品检查、有效期标注、操作验证、差异记录及重新包装要求等多个方面的检查。

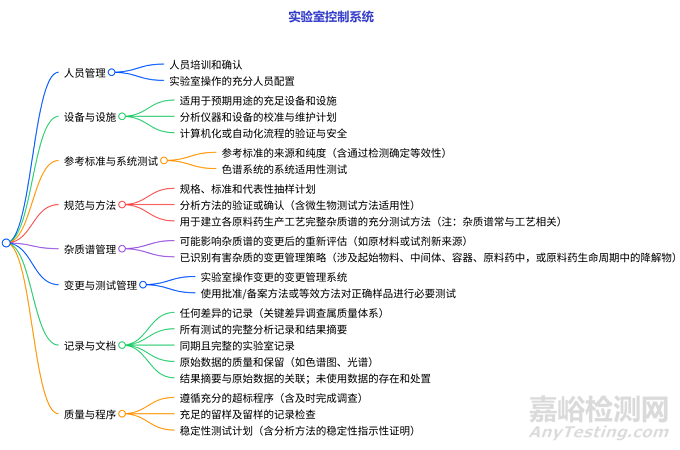
实验室控制系统的检查
包含人员管理、设备与设施(配置、校准维护、自动化流程安全)、参考标准与系统测试(来源纯度、色谱系统适用性)、规范与方法(抽样计划、方法验证、杂质谱测试)、杂质谱管理(变更后重评估、有害杂质变更策略)、变更与测试管理(操作变更系统、样品测试要求)、记录与文件(差异记录、分析记录等)、质量与程序(超标处理、留样、稳定性测试)等多方面的检查,明确了需要评估的内容及相关要点。

来源:GMP办公室


