您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2023-07-24 11:17
本文适用于通过改变体位、起立角度对患者进行训练促进康复的康复训练床,该类产品通常含有电动控制装置,用于对脑中风、脑外伤等患者进行肢体运动康复训练。
该类产品在《医疗器械分类目录》中分类编码为19-02-02。 产品的管理类别为Ⅱ类。
一、康复训练床的工作原理和结构组成
1、工作原理
直立康复训练床是通过调整倾斜角度使被缚于其上的患者产生自身重力作用,帮助患者完成仰卧位到站立位,重心从低到高的过渡,使患者充分适应立位状态。提高躯干和下肢的负重能力,增加颈、胸、腰及骨盆在立位状态下的控制能力,为将来的自主立位及平衡的保持打下良好的基础。
多体位康复训练床有两块以上床板,通过电动控制装置可以独立调节各床面的高度和角度,通过机械支撑系统支撑训练床自身以及床上患者的升降,促使床体高度及床面各段位不同 角度的调节得到简化。训练位置能得到充分的调整,辅助使用者进行多种姿势训练。
PT康复训练床是采用电机、控制盒及脚控开关等进行调节控制的康复训练床。可根据康复患者的需要,对床面高度进行调节,患者配合治疗师进行体位调整,方便治疗师对患者全身 部位进行诊断、检查、治疗和按摩。
2、结构组成
PT 康复训练床通常由床板、机械支撑部件、电动控制装置、 固定保护装置、脚轮等组成。具体产品结构示意图见图1。
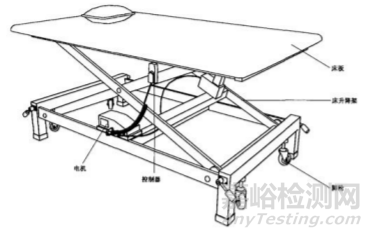
图1 PT 康复训练床 直立康复训练床通常由床板、机械支撑部件、电动控制装 置、固定保护装置、扶手、脚轮、脚托板等组成。具体产品结构 示意图见图2。
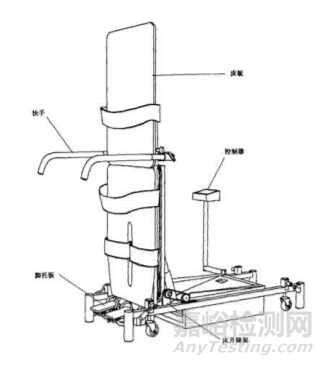
图2 直立康复训练床 多体位康复训练床通常由床板、机械支撑部件、电动控制装 置、脚轮等组成。具体产品结构示意图见图3。
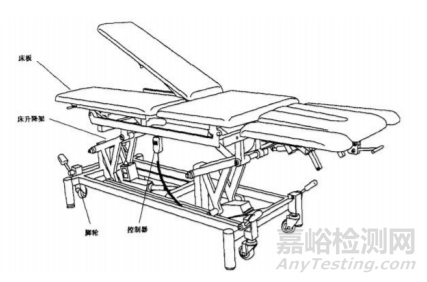
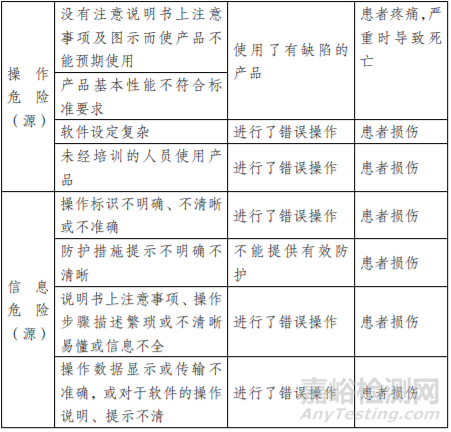
二、康复训练床的主要风险
应按照YY/T 0316-2016 附录C 的34 条提示对康复训练床产品的安全特征进行判定,并按照YY/T 0316-2016 附录E 的提示,通过对产品的危害、可预见事件序列和危害处境进行全面分析和评价,并有针对性地实施降低风险的技术和管理方面的措施。
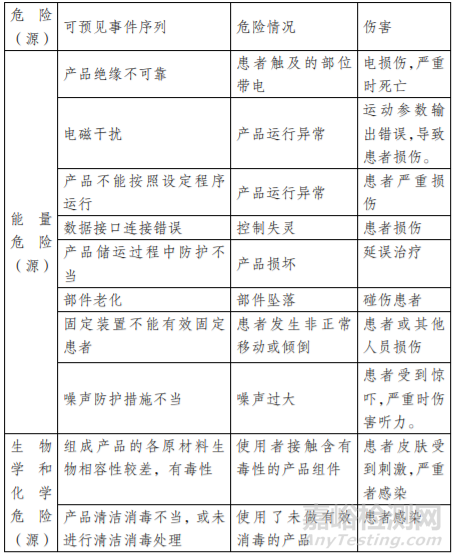
以下给出了康复训练床常见的风险要素及示例(如表 1)。 由于产品的工作原理、结构组成、性能指标存在差异,所以这些风险要素并不是全部,开发人还应根据产品特点确定其他风险 并进行有效控制。
表 1:危险(源)、可预见事件序列、危险情况及伤害示例
三、康复训练床的产品性能研究实验要求
1、性能指标和安全要求
康复训练床应当符合GB/T 26340《可调式康复训练床》 的要求。
配有脚踏开关的设备,应符合YY 1057《医用脚踏开关通 用技术要求》的要求。
若提供心率、血压、脉搏等生理指标监测功能,应符合相应标准的要求。
软件:若适用,应按照《医疗器械软件注册技术审查指导原则》(2022 年修订版)(2022 年第9 号)制定软件性能指标。 “性能指标”包括软件的功能、使用限制、接口、访问控制、运行环境(若适用)、性能效率(若适用)等要求。
电气安全应当符合GB 9706.1《医用电气设备 第1部分: 安全通用要求》的要求。
电磁兼容应符合YY 0505(YY 9706.102)《医用电气设备 第1-2 部分 安全通用要求并列标准 电磁兼容 要求和试验》的要求。
康复训练床应符合GB 24436《康复训练器械 安全通用 要求》(5.10 电器安全除外)和GB/T 26340《可调式康复训练床》的要求。
2、化学和物理性能研究
应当明确产品化学/材料表征、物理和/或机械性能指标的确定依据、设计输入来源以及临床意义,所采用的标准或方法、采用的原因及理论基础。
2.1功能性指标的验证可参考GB/T 26340-2010 的要求,至少应包括以下指标:活动部件、面边角和管端、结构设计、稳定性、机械强 度、线性尺寸与角度要求、操作力、操作速度和时间、脚轮定位、 噪音、控制器、操作手柄和踏板、电源软电线等。直立床还应包括附件及强度要求,附加稳定性要求。
2.2安全性指标的验证包括电气安全指标和电磁兼容指标两大类。安全指标应当包括GB 9706.1、GB 24436(5.10 电器安全 条款除外)及其他适用的国家标准和行业标准中的所有指标,电磁兼容指标应当包括YY 0505(YY 9706.102)及其他适用的国家标准和行业标准中的所有指标。
3、电气系统安全性研究
应当开展电气安全性、机械和环境保护以及电磁兼容性的研究,明确适用的标准。
4、软件研究
应按照《医疗器械软件注册技术审查指导原则》 (2022 年修订版)(2022 年第9 号)的要求开展软件相关研究。 如为自研软件,应开展自研软件研究。如为现成软件,应开展现成软件组件研究。
如产品适用于医疗器械网络安全,包括具备电子数据交换、远程访问与控制、用户访问三种功能当中一种及以上功能,应按照《医疗器械网络安全注册技术审查指导原则》(2022 年修订版)(2022 年第7 号),开展医疗器械网络安全研究。
5、生物学特性研究
生物学相容性评价应根据产品与人体接触部位、接触方式及接触时间,按GB/T16886.1-2011《医疗器械生物学评价 第1 部分:风险管理过程中的评价与试验》的要求进行。
生物相容性评价研究应关注:
(1)生物相容性评价的依据和方法。
(2)产品所用材料及与人体接触的性质。
(3)实施或豁免生物学试验的理由。
(4)对于现有数据或试验结果的评价。
依据GB/T 16886.1 附录A《生物学评价试验》中表A1 要考虑的评价试验,需要做的生物相容性评价试验为细胞毒性、 迟发型超敏反应、皮内反应或刺激。
可根据《关于印发医疗器械生物学评价和审查指南的通知》 (国食药监械〔2007〕345 号)进行生物学评价。
生物学评价主要对以下内容进行评价:
(1)医疗器械材料的定性与定量的说明或分析。
(2)医疗器械/材料与市售产品的等同性比较:比较材料和产品的用途是否等同;比较两者的生产过程(加工过程、灭菌过程、包装等)是否相同。
生物性能试验要求应关注以下内容:
(1)细胞毒性试验:按照GB/T 16886.5 中规定的方法进行检验,应≤2 级(定性评价)或无潜在毒性影响(定量评价)。
(2)皮内反应或刺激:按照GB/T 16886.10 中规定的方法进行检验,极性浸提和非极性浸提下应无皮内刺激反应。
(3)迟发型超敏反应试验:按照GB/T 16886.10 中规定的 方法进行检验,极性浸提和非极性浸提下应无迟发型超敏反应。
材料名称相同并不代表材料相同,除非来自同一供应商(即材料的生产商,而非经销商或代理商)。不同的供应商材料的配方可能存在差异。
6、清洁、消毒、灭菌研究
6.1生产企业灭菌:应明确灭菌工艺(包括灭菌方式和相关参数)和无菌保证水平(SAL),并开展灭菌确认。
6.2终端用户灭菌:企业应当明确推荐的灭菌工艺(包括灭菌 方式和相关参数)及所推荐的灭菌方法确定的依据;对可耐受 两次或多次灭菌的产品,应当开展产品相关推荐的灭菌方法耐受性的研究。
6.3残留毒性:如灭菌使用的方法容易出现残留,应当明确残留物信息及采取的处理方法,并开展相关研究。
6.4终端用户消毒:应当明确推荐的消毒工艺(方法和参数) 以及所推荐消毒方法确定的依据。
7、产品稳定性研究
7.1货架有效期
如适用,应当开展货架有效期和包装研究,证明在货架有效期内,在生产企业规定的运输贮存条件下,产品可保持性能功能满足使用要求,具有微生物限度要求的产品还应当符合微生物限度要求,以无菌状态交付的产品还应保持无菌状态。
7.2使用稳定性
如适用,应当开展使用稳定性/可靠性研究,证明在生产企业规定的使用期限/使用次数内,在正常使用、维护和校准(如适用)情况下,产品的性能功能满足使用要求。
7.3运输稳定性
应当开展运输稳定性和包装研究,证明在生产企业规定的运输条件下,运输过程中的环境条件(例如:震动、振动、温度和湿度的波动)不会对医疗器械的特性和性能,包括完整性和清洁度,造成不利影响。

来源:嘉峪检测网


