您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2023-05-22 11:11
|
试验项目 |
目的 |
|
|
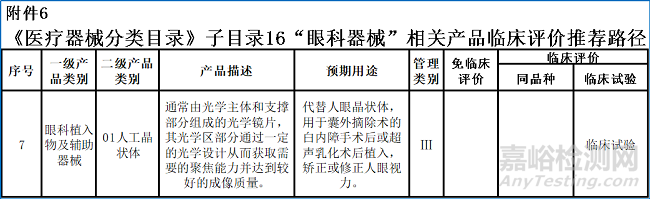
1 |
极限浸提(附录A) |
确定并量化人工晶状体中可浸提材料的总量, 合成过程和添加剂中可能存在的残留物, 或生产和包装过程中的杂质,并用来进行风险评定。 |
|
2 |
可沥滤物(附录B) |
确定并量化人工晶状体在模拟生理环境下释放的物质, 用来确定临床使用过程中的风险。 |
|
3 |
水解稳定性(附录C) |
确定并量化因水解作用可能产生的降解产物, 以确定人工晶状体在水性环境下的稳定性, 并分析水解产物的潜在风险。 |
|
4 |
抗紫外线/可见光的光照稳定性 (附录D) |
表述紫外线/可见光照射对人工晶状体光学、 机械和化学性能的影响, 并分析光照产物的潜在风险。 |
|
5 |
Nd-YAG激光照射稳定性 (附录E) |
确定人工晶状体在Nd-YAG激光照射对 人工晶状体化学性能的影响, 并分析激光照射产物的潜在风险。 |
|
6 |
不溶无机物 (残留物水平及风险评价) |
量化在生产加工和包装过程中的不溶无机物含量, 并进行风险分析。 |
|
试验项目 |
参考标准 |
试验要求 |
|
|
1 |
致敏 试验 |
ISO 10993-10/ GB/T 16886.10 |
可选用最大剂量试验方法。 局部淋巴结分析试验(LLNA)可在有充分证明的情况下使用; 试验材料应采用两种不同的浸提介质,一种为生理盐水, 另一种为脂溶性或偶极性溶剂。 脂溶性或偶极性溶剂应不溶解或降解试验材料。 溶剂本身不是已知的刺激物、辅助剂或致敏物。 |
|
2 |
遗传 毒性 试验 |
ISO 10993-3/ GB/T 16886.3 |
应对材料进行两种溶剂的浸提,一种为生理盐水, 另一种为脂溶性或偶极性溶剂。脂溶性或偶极性溶剂应不溶解或降解试验材料。 应在37 ℃± 2℃,72 h ± 2 h条件下进行浸提。 当遗传毒性试验需要使用浸提液进行额外稀释时, 稀释系数应在选定的浸提比例范围内。 注:按ISO 10993-3/GB/T 16886.3进行遗传毒性试验要使用一个试验组合, 因为一个单独的遗传毒性试验不能说明所有的遗传毒性风险。 |
|
3 |
局部 反应 试验 |
ISO 10993-6/ GB/T 16886.6 & ISO 11979-5/ YY/T 0290.5 |
按ISO 10993-6/GB/T 16886.6和ISO 11979-5/YY 0290.5 附录F的补充要求进行植入后局部反应试验。 |
|
4 |
眼 植入 试验 |
ISO 10993-6/ GB/T 16886.6 |
若制造商不能提供人工晶状体在眼内环境下材料的安全性的文件证明, 应进行眼植入试验。该试验应按ISO 10993-6/GB/T 16886.6进行, 并补充ISO 11979-5/YY 0290.5附录G的要求。 当认为该试验不需要时,其风险评定应提供合理的保证, 根据以前的临床使用信息和其他相关文献, 确保试验材料的新用途所产生的风险是可接受的 |


来源:微谱医疗器械技术服务


