强降解试验的目的并非是为了实现分析结果的质量平衡,而是对降解化学有一个比较全面的了解:1)了解药物的降解路径及分子内稳定性;2)建立稳定性指示分析方法,使其适用于样品检测;3)为药品的处方、工艺、包装、贮藏条件的确定提供有益支持,以便于稳定性试验的顺利进行。在一些情况下,降解产物(杂质)质量(或摩尔数)的增加小于母体化合物(主成分)质量(或摩尔数)相应的减少。问题的潜在来源和解决方法如下所述:
(一)降解产物在色谱柱上未被洗脱
假定母体化合物和所有降解产物都是完全可溶的,并且可以通过HPLC-UV检测。有以下方法可以诊断此问题:
(a)可以修改HPLC方法以洗脱其他杂质
可以通过增加流动相的强度(即增大有机相比例)或增加分析时间来修改HPLC方法,以洗脱保留较强的非极性化合物。
(b)可以使用紫外分光光度法将部分降解的样品与未降解进行分析,并将结果与通过HPLC分析获得的结果进行比较。
此方法对于使用UV检测器的HPLC方法很有用。由于紫外分光光度法不涉及分离,因此不会由于化合物保留在色谱柱上而被遗漏。使用这种方法,可以将部分降解的样品在流动相中溶解或稀释。获得部分降解样品完整的UV(或VIS)光谱,并将其与未降解样品的光谱进行比较。在HPLC方法中使用的波长下获得部分降解样品与未降解样品的吸光度之比。然后将该吸光度比与通过HPLC方法获得的部分降解样品与未降解样品的总峰面积之比进行比较。如果使用HPLC方法检测到所有杂质,则部分降解样品的总HPLC峰面积除以未降解样品的HPLC峰面积应等于吸光度比。如果HPLC方法利用光电二极管阵列(PDA)检测器,则可以根据需要在多个波长下确定比较。如果HPLC面积比明显小于分光光度法吸光度比,则存在一种或多种降解产物没有从色谱柱上洗脱。
(c)可以使用不接色谱柱的HPLC系统分析样品(即流动注射分析)。
在该实验中,用双通代替HPLC系统中的色谱柱,将获得的总峰面积与使用色谱柱时获得的总峰面积进行比较。如果在接色谱柱时获得的总峰面积明显小于不接色谱柱时的总峰面积,则HPLC方法中存在一些杂质未被洗脱。在不接色谱柱的情况下,所有杂质和主成分共同洗脱,得到一个未保留的峰,其面积与可检测物质的总量成正比例。假设所有物质都被洗脱,则色谱柱存在时所有峰的总峰面积应相同。如果色谱柱存在时与不接色谱柱时总峰面积差异较大,则在给定的色谱条件下很可能一种或多种化合物没有被洗脱。这种诊断方式的潜在困难是易受溶解样品溶剂的影响,若溶剂与流动相有很大不同,则溶剂对样品响应的影响可能会使在没有色谱柱的情况下难以进行准确的积分。此外,进样量产生的峰面积不能超出线性范围。最后,如果在使用色谱柱时使用了流动相梯度,则不断变化的溶剂组成会影响分析物的响应,从而影响洗脱物质的峰面积。当梯度流动相组成的每个极端值均等地使用时(等度),可以通过比较不使用色谱柱而获得的总面积来确定梯度是否具有这种影响。
(二)检测器未检测到降解产物
HPLC-UV是最常用的检测技术。尽管广泛适用,但紫外检测器不能检测所有化合物。降解可能会产生没有生色团的化合物,在这种情况下,观察到的降解产物增加量将小于母体化合物的损失量。该问题的诊断可使用更低的波长或使用其他检测器(例如,蒸发光散射检测(ELSD),带电气溶胶检测(CAD),质谱法(MS)或火焰电离检测(FID)。必须牢记的是,虽然此类检测器适用范围广,但与UV一样,其响应也不均匀。例如,蒸气压明显的化合物通过ELSD和CAD给出的响应较差,而MS响应随可电离性变化很大。但是这种检测器对于确认原始方法未检测到的降解产物的存在可能非常有用。
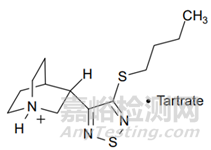
检测器未检测到降解产物的示例如候选药物LY297802。LY297802水溶液在冷白色荧光灯(约17000 lux)下破坏3天,质量损失了5.4%;破坏7天,质量损失了42%。但是,通过HPLC-UV有关物质方法(梯度方法),降解产物的增加量分别为0.4%和1.3%,明显存在分析质量不平衡。仔细检查盛有样品的容器,发现表面沉积了不溶性雾状薄膜。收集该不溶性薄膜并通过EI-MS分析,表明该薄膜含有元素硫。由于硫很可能源自噻二唑环的降解,考虑生成没有生色团化合物的可能性,及形成挥发性产物的可能性。用己烷萃取(从轻度降解的水溶液中碱化,得到LY297802的游离碱),然后进行GC-FID分析,发现降解样品中有两种主要的降解产物。使用GC-MS进行分析,提供了分子式信息,阐明了结构,提供了对降解化学的了解以及HPLC-UV缺乏可检测性的原因(如,降解产物易挥发且无紫外吸收)。
图1 LY297802结构
在某些情况下,由于不溶性,挥发性或吸附损失,降解产物会无意中从检测样品中排除。通常,最难解决的问题是最明显且最直接的解决方法。在这种情况下,目视观察或浊度检测可能会发现问题所在。例如,在较早描述的化合物LY297802的降解溶液中,观察到一些不溶性物质,并确定其为元素硫,为质量平衡问题提供了有价值的线索。当然,当存在于含有不溶性辅料的药品中时,不溶性降解产物不太明显。在这种情况下,与安慰剂或未降解的样品相比,不溶性化合物的分离和检查是合适的。
如果降解产物由于其挥发性而损失,则可以使用其他分析技术。在这种情况下,使用溶剂-溶剂提取样品可能是合适的。萃取(如前面提到的LY297802示例中所述)或以顶空方式捕获降解样品。然后可以使用气相色谱法(GC-FID或GC-MS)比较降解和未降解样品的结果。在有限的情况下,如果降解是由热引起且迅速发生,则TGA和相关的气相色谱分析可能是一种有用的方法。最后,如果降解可以在低温下产生,则可以使样品保持低温以最小化挥发性。
降解产物也可能吸附在样品容器或不溶性辅料中,在后一种情况下,问题的诊断和解决方法与降解产物不溶性相同(参见上文)。如果吸附到容器上是一个潜在的问题,那么最直接的方法是使用不同的容器材料(例如玻璃和聚丙烯)比较结果。在某些情况下,可能需要对容器表面进行去活或更改样品溶剂(例如pH和溶剂强度)以最大程度地减少吸附。
(三)母体化合物在样品基质中丢失
在极少数情况下,由于挥发性或吸附性,母体化合物本身可能会从样品基质中流失。通常,即使发生此类损失,与母体化合物的质量相比也微不足道。但是,如果有意义的话,测定结果的下降将不会是由于降解引起的,因此不会对应于降解产物的任何增加。通常,在强降解试验之前获得的信息(例如,熔点、沸点,蒸气压和吸附到各种材料上的趋势)应提供此类潜在问题的迹象。如上所述进行诊断和解决。
(四)在有关物质方法中与母体化合物共洗脱的降解产物
如果在有关物质方法中降解产物与母体化合物共洗脱,那么在给定条件下,所得质量平衡取决于杂质相对于母体化合物的响应因子。如果所形成的杂质具有较小的响应因子,则存在质量平衡为正。如果杂质的响应因子大于母体的响应因子,那么质量平衡将为负。如果响应因子相同,则不会影响质量平衡。通过光电二极管阵列(PDA)-紫外检测或通过MS检测可以揭示共洗脱杂质为峰异质性。当然,PDA检测仅对具有不同UV光谱且不能与母体完美共洗脱的杂质有效。另外,PDA对检测峰异质性的敏感性取决于杂质的光谱特性与母体有多大差异。对于某些杂质,PDA检测器可能无法检测到含量低于百分之几的杂质。LC-MS可以对广泛的潜在共洗脱杂质提供更高的灵敏度。然而,LC-MS更昂贵,可以使用的流动相类型有限。也可使用正交分离技术(例如,CE)来检查共洗脱杂质。如果发现共洗脱问题,应修改有关物质方法以分离出共同洗脱的杂质。
(五)色谱性能不良导致降解产物未积分
一些降解产物的色谱性能可能不佳例如由于与痕量金属杂质或残留硅醇基的不良相互作用,在柱上从一种产物转化为另一种产物等,尽管它们可能从HPLC色谱柱上洗脱下来,但所得色谱峰变宽了(通常严重拖尾),很容易被“遗漏”并保持未积分状态,尤其是当宽峰且峰面积较低时。如果母体化合物降解为溶解性差的杂质,则分析后观察到的色谱图可能不会显示出离散的峰,而是基线升高。在低水平下,这样高的基线很容易被遗漏。诸如此类的情况并不少见,特别是在使用等度HPLC方法的情况下。运行空白对于确定基线升高是来自样品还是属于色谱假象,非常有帮助。确定质量平衡结果是否来自不良的色谱性能的实验与确定降解产物是否未从色谱柱中洗脱的实验相同(请参见“未从HPLC色谱柱中洗脱的降解产物”一节)。
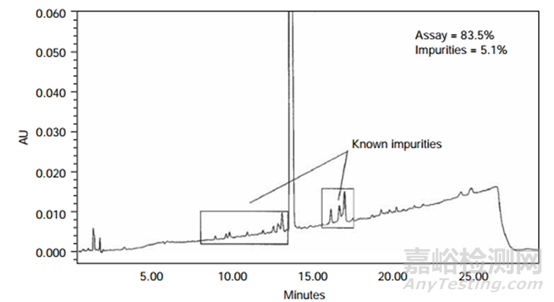
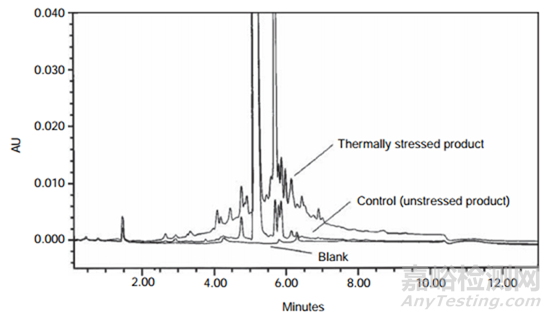
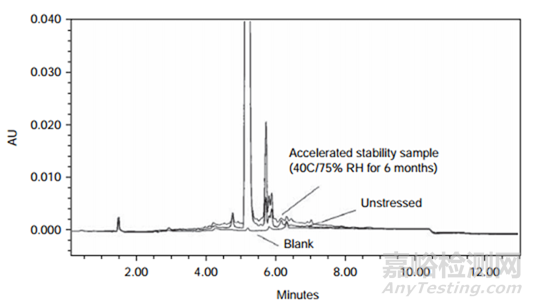
母体化合物降解产物之间分离度较差的示例如图1所示。在本例中,通过RP-HPLC分析了部分降解的样品(在25分钟内,乙腈的梯度为5-70%),并且检测到许多具有已知响应因子的已知降解产物,但是质量平衡为88.6%。考虑了降解产物未从色谱柱上洗脱下来的可能性,并设计了一个实验来检验该假设。在该实验中,在没有接色谱柱的情况下分析了降解样品(流动注射)。如果使用RP-HPLC方法检测到所有降解产物,则HPLC含量和有关物质结果的总和(即83.5 + 5.1 = 88.6%)应等于流动注射结果。流动注射结果为97.3%,表明使用RP-HPLC方法未检测到大量的降解产物。该差异与降解产物未从色谱柱上洗脱出来或未进行积分相一致。为了研究产物是否未从色谱柱上洗脱,对梯度RP-HPLC方法进行了改进,将乙腈的比例提高至90%,并在此比例下保持1小时,但并未洗脱出任何其他降解产物,这表明没有任何强保留的非极性降解产物。为了测试降解产物色谱性能不佳的可能性,进行了另一项实验。在该实验中,使用等度HPLC方法对样品进行了测定,该方法有机相比例较高,以单峰洗脱母体化合物和降解产物。获得的结果与流动注射分析的结果相当,这表明所有降解产物都从色谱柱上洗脱下来。该发现表明色谱分离不良的降解产物未与原始RP-HPLC方法积分在一起。通过将原始RP-HPLC梯度修改为陡峭梯度(在25分钟内使用0-90%乙腈),证实了这一假设。如图3所示,比较了未破坏样品和降解样品(质量平衡较差)。注意基线大约在3-8分钟升高,仅在破坏样品上可见,而在对照样品上看不到。将该基线“峰”的积分并包括在总峰面积(母体和总杂)中,得到的结果与使用流动注射分析获得的结果相似。当色谱图和空白的色谱图重叠时,在降解不那么严重的样品中也可以识别出该基线峰(如图4所示)。因此,低的质量平衡归因于色谱性能差的物种,这些杂质以前没有被积分,因为它们被忽略为仅仅是基线波动。
图2 使用梯度HPLC在高温破坏样品上获得的HPLC色谱图
图3使用陡梯度HPLC方法在高温破坏样品,未破坏样品和空白样品上获得的HPLC色谱图叠加图
图4 使用陡梯度HPLC方法分析6个月加速稳定性条件下的样品,未破坏的样品和空白所获得的叠加HPLC色谱图
(六)响应因子差异导致定量不准确
如果未校正的峰面积代表实际相对量,杂质和母体化合物之间的响应因子(例如,给定波长下的吸收率)显著不同,则质量不守恒是不可避免的。如果降解产物的响应因子小于母体化合物的响应因子,则会导致质量平衡为正。当降解产物具有较大的响应因子时,将导致质量平衡为负。因此,杂质的相对响应因子(RRF)是评估质量平衡时的重要考虑因素。
有时,可以合理地假设UV响应因子非常相似。例如,如果母体和降解产物共享相同的生色主链,则仅在与生色团无关的区域具有结构差异。UV光谱的有利比较(例如,来自PDA检测器的光谱)可以帮助确认这种情况。
通常,建立响应因子的过程需要使用单个杂质的对照品。为了准确测定,必须知道杂质的纯度,然后根据峰面积及浓度比计算响应因子。但是要综合考虑杂质与母体化合物之间的紫外吸收光谱图,选择合适的紫外吸收波长,否则,也会导致质量不守恒。
因此,在强制降解试验前需考虑样品的理化质,了解药物在各个条件下可能的降解机制以及降解反应的难易程度,可能形成的降解产物,合理的设计试验,以确定合适的降解条件。
参考文献:
[1] Pharmaceutical Stress Testing Predicting Drug DegradationSecond Edition.Steven W.Baertschi,Karen M. Alsante,Robert A. Reed。