您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-05-14 14:49
摘要
自1998年第一份混合均匀性指导原则开始,关于药物混合均匀性取样方法以及接受标准的讨论就从未停止过,随着讨论的不断深入工业界对产品的质量控制亦走向越来越科学的方向。
FDA初代指导原则
1999年8月,美国食品药品监督管理局 (FDA) 发布了一份名为“Guidance for Industry, ANDAs: Blend Uniformity Analysis”的指导原则, 专门用于指导ANDA 产品的开发,其中详细的列出了混合均匀度抽样和验收标准,然NDA的亦同样用该方法进行混合均匀度的评价。该方法是独立于USP成品放行之外的过程检测方法,并在其中提到只要产品不符合混合均匀性要求无论最终产品是否符合含量均匀度要求该批产品最终都不得放行。产生上述矛盾的根本问题在于混合取样问题(取样器相关的混合采样错误问题)和分析方法不足(混合样品分析过程中的称重误差),然该指导原则中提到的混合均匀度限度依然被国内众多药企所奉行。
PQRI脑洞大开
通常而言,最终制剂的含量均匀性很大程度上取决于混合机内粉体的均匀性,然对于存在潜在分层风险的物料而言混合器内的均匀性则很难确保最终制剂的含量均匀性。基于99指导原则中过程检测和放行测试矛盾问题,2000年产品质量研究院(PQRI混合均匀性工作组(BUWG)) 就ANDA和NDA向FDA提交了一份提案,建议对采用最终混合取样和过程中剂量单位使用分层抽样相结合的方法来评价混合均匀性,99指导原则与2002年被FDA召回。次年FDA发布了“Guidance for Industry :Powder Blends and Finished Dosage Units—Stratified In-Process Dosage Unit Sampling and Assessment” 由此开启了以过程分层取样来评价混合均匀性的重要跨越。分层取样评估混合均匀性优势主要体现在如下几个方面:
1.该方法可以对产品的同质性进行精确的度量。
2.该方法消除了与取样器采样相关的错误问题。
3.该方法提供了可靠、准确的产品质量信息。
4. 该方法消除了混合样品分析过程中的称重误差。
5. 该方法消除了在隔离环境中生产的有毒或强效药物混合取样的安全问题。
6. 该方法可以解释了混合后物料在压片或灌装过程中潜在分离行为。
分层取样检测的方法对后续控制策略产生了显著的影响,下图可以很好的说明03版指导原则的关键性突破。
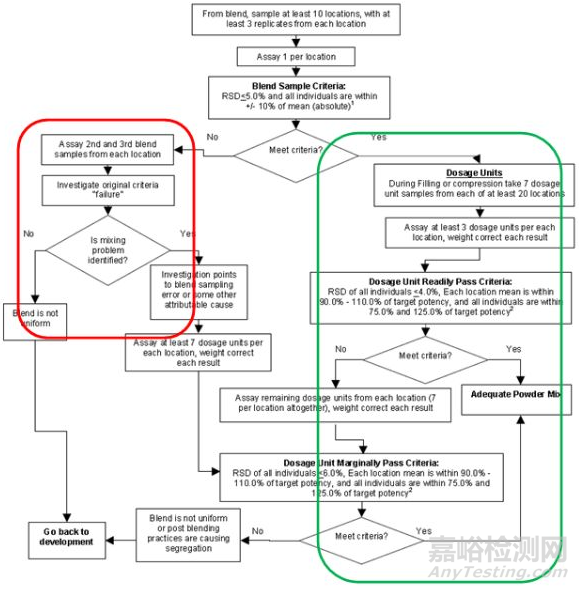
该指导原则测试方案流程如下:
1.混合器内粉体测试
1.1 至少10个具有代表性的不同取样位置,每个位置三份样品,每份样品1-3倍制剂当量(超出3倍则需要合理解释)
1.2 每个位置测量一份样品,若RSD≤5.0且单点值在均值±10%范围(例如均值为95%,则范围为85%-105%,而非95%±9.5%)之内则通过测试,进行下一工序开始分层取样(若第一份未通过测试,测试第二份和第三份,根据调查结果确定是否进入下一工序)
2.分层取样测试
2.1 分层取样20个时间位置点,每个点至少取样7个单位,每个点测试3个单位(单点结果进行片重校正),若:20*3的RSD≤6.0,且20个取样位置点三份均值在90%-110%且单点含量值在75%-125%,则可判定为混合均匀,反之每个点需要额外至少测4份并满足上述条件则可判定混合均匀。
上图绿色区域则是分层取样评价粉体混合均匀度的核心所在,增加过程分层取样点(20*7),相较于99版指导原则若产品混合均匀度不满足红色区域时该批产品则视为失败批而言2003版本则给予这种现象一个进一步判断的空间,即表明该指导原则侧重于以分层取样数据来最终判断粉体是否混合均匀。
2003版指导原则中建议在产品展示和工艺验证批次的生产过程中采用混合器内取样和过程中剂量单位分层抽样相结合的方法,以证明共混物的均匀性,方法如下。
混合器内取样:在混合器内至少选择10个取样位置进行取样,每个位置至少取3份样品。取样位置必须仔细选择以代表混合不良行为发生的潜在区域。
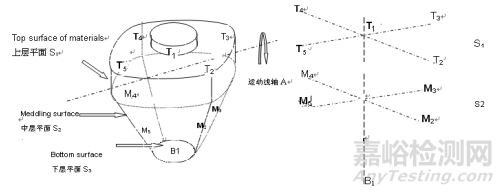
例如,在翻滚搅拌机(如v型搅拌机、双锥搅拌机或滚筒搅拌机)中,应从搅拌机轴线上至少2个深度处选择样品。对于对流混合器(如带状混合器),应特别注意取样位置和取样量的统一,包括拐角和排放区域(建议至少20个位置,以充分验证对流混合器)。
过程分层取样:在整个压片或填充操作过程中,确定至少20个取样点。采样点必须仔细选择,以覆盖压片或填充过程中的重大事件(例如料斗的转换),包括压片或填充操作开始和结束时的样本,每个部位至少取7个剂量单位。
ISPE优化升级
2013年8月服役10年之久的“Guidance for Industry :Powder Blends and Finished Dosage Units—Stratified In-Process Dosage Unit Sampling and Assessment”退出历史舞台,主要原因在于03版本指导原则虽然在混合器内取样时已经关注了取样点的重复性但未对同一取样点的均匀性进行分析于此同时虽然采用了重复样品但在混合均匀性评估时仅仅用了其中一组数据。FDA则认为应对所有的不同位置的重复取样样品均应进行统计学分析,该统计学分析可证明粉体是均匀的同时取样位置的变动对检测结果不产生显著性影响。
基于上述考虑ISPE(国际制药工程协会)于2014年在03版的指导原则之上修订产生了“Recommendations for the Assessment of Blend and Content Uniformity: Modifications to Withdrawn FDA Draft Stratified Sampling Guidance”。相较于03版指导原则该建议主要优化了混合器内样品混合均匀性可接标准,并初步提出基于混合器内物料混合均匀性、过程分层取样均匀性以及终产品含量均匀度三者数据之间的关联性尝试建立以分层取样数据作为终产品含量均匀度放行的重要思考。
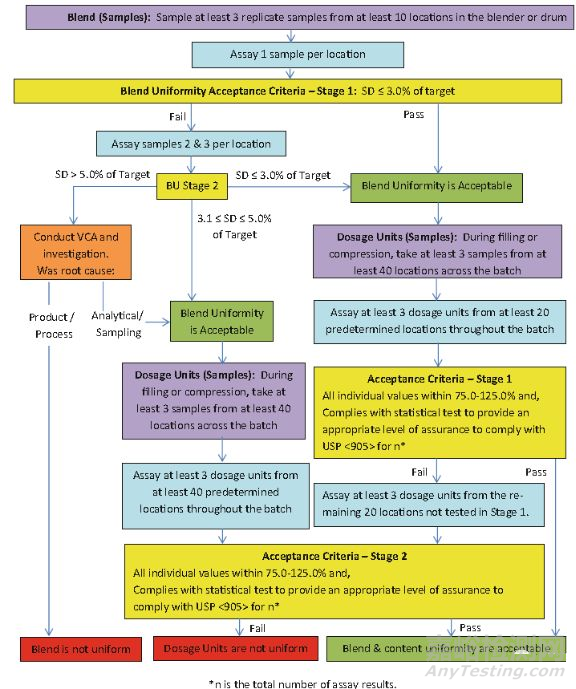
如上图所示,ISPE的取样手法依然保留了03版的内容,其中更新了对于混合器内混合均匀性样品进行BU进行了 Stage2 (30个数据分析)的判断并提高了Stage1的判断限度5.0%→3.0%,并增加了分层取样点密度(20个点→40个点)同时在判定标准中取消了重量校正的操作,直接以单点值(20*3)进行结果判定这样以来该分层取样数据就能和放行的含量均匀度数据建立直接的相关性为后来在线放行的理念做了很好的铺垫。
ASTM开启统计时代
伴随统计学在制药领域的不断应用以及PAT技术的不断推广,粉体混合均匀性判断也迎来属于它的统计分析时代,FDA在废止03版本是就以提及General Chapter <905>Uniformity of Dosage Units of the USP 并不是一个科学的测试方案,该结果亦不能用于外推到大量样品以及不能评估取样点内部的均匀性问题。
基于此ASTM 推出ASTM E2709“Standard Practice for Demonstrating Capability to Comply with an Acceptance Procedure”和 ASTM E2810“Standard Practice for Demonstrating Capability to Comply with the Test for Uniformity of Dosage Units”ASTM 在取样操作上保留03指导原则的基础上增加了取样量、检测量的灵活性,操作者基于对自己产品的认知制定灵活的取样测试方案。
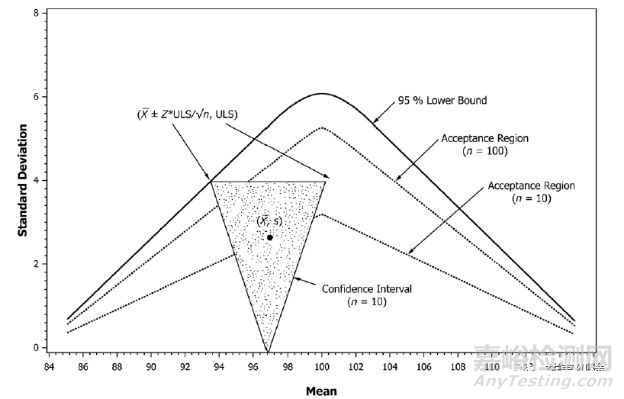
如上图所示,ASTM E2810主要基于样本量的均值以及SD值、置信水平以及概率界限来生成可接受限度表。上图则表示在已知样本量均值以及SD的条件下,95%概率曲线覆盖范围内的数据即可通过含量均匀度测试。
基于产品质量过程控制的认识,过程控制标准往往高于放行测试标准,以含量均匀度为例(假设均值X=100%,n=10或者30时)分别进行<905>和E2810限度判定如下
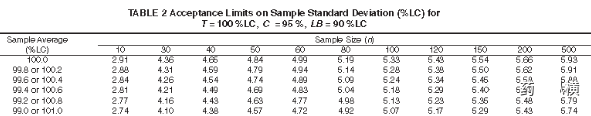

放行测试是n=10时SD≤6.25(15/2.4)、n=30时SD≤7.5(15/2.4)即可,而在过程分层取样中n=10的限度仅仅为2.91、n=30的限度仅为4.36(LB=90%LC条件下),由此可见基于过程分层取样控制限度比放行测试允许限度更为严格。
文末杂谈
笔者对于2709和2810中相关统计学研究比较浅陋,仅处于简单判断层面的理解。然从整个的指导原则发展过程可知制药工业的在线控制或者过程控制已经发生了翻天覆地的变化,笔者经常在丁香园等专业论坛看见诸多战友因BU取样问题导致的结果不理想而困惑,才发现我们很多时候对混合均匀性和含量均匀度的判断依然停留在过去式,就笔者本身而言在过往的项目开发中对此也是重视不够,对最终结果的判断也是缺乏科学根据,更多时参考前人留下经验方法,更有甚时为了所谓的BU检测合格将取样量一度提高到10倍的单位剂量。
分层取样的操作在03版指导原则中建议在展示批(注册批或工艺验证批)进行、在生产批也应持续验证,然目前我们很多企业在该方面的工作依然是相对薄弱的,当然导致这种现象的原因是多方面的,一方面我们目前在产品转移过程中很多时候是注重一次性的成功并未制定详细的产品控制策略,另一方面对于该方面的重视不足,研究阶段由于批量限制一般未进行分层取样考察,工艺转移或者放大过程中过程取样很多时候是为了确认制剂单元重量的一致性并未采用单元含量测定来确认混合工艺的均匀性和制剂含量均匀度。
于是查阅发现国内相关的技术文件相对单薄仅有一份GBT5918-2008可参考其中也对CV值进行控制(cv不得大于5%)。基于上述思考以及目前对该点判断的诸多困惑,笔者对混合均匀性的历史做此简文叙述以冀各位同行在对此引起关注进而优化药品制造过程中的质量控制策略。
参考文献:
1. Evaluation of blend uniformity and content uniformity based on 2003 stratified sampling guidance and 1999 blend uniformity analysis guidance: product A.
2. Guidance for Industry :Powder Blends and Finished Dosage Units—Stratified In-Process Dosage Unit Sampling and Assessment
3. Recommendations for the Assessment of Blend and Content Uniformity: Modifications to Withdrawn FDA Draft Stratified Sampling Guidance
4. Assessment of Blend and Content Uniformity. Technical Discussion of Sampling Plans and Application of ASTM E2709/E2810

来源:药事纵横


