摘要
目的:对美国食品和药物管理局(FDA)发布的药物非临床研究警告信进行分析,为药物非临床研究质量管理规范(GLP)监管提供参考。
方法:收集2008年至2022年FDA对非临床研究发出的警告信,统计警告信数量及问题分布情况,解析和归纳警告信中公布的研究者在非临床研究中发生的主要问题。
结果:FDA对非临床研究发布的12封警告信中共有67项问题数,涉及21CFR58条款120项次。其中发生频率最高的缺陷是质量保证部门人员履职不足、专题负责人履职不足和研究过程中方案的实施及报告撰写。
结论:FDA对药物非临床研究警告信中高频率问题集中于项目负责人及质量保证部门的履职尽责方面。监管机构应在人员的履职尽责方面重点关注,促进非临床研究质量的提升。
关键词
药物非临床研究质量管理规范;警告信;美国食品和药物管理局;研究人员
药物非临床研究是为评价药物安全性,在实验室条件下用实验系统进行的实验,是药物研发的基础工作,也是临床试验的重要基础数据,被认为是人体试验前的最后一道防线[1]。药物非临床研究质量管理规范(non-clinical good laboratory practice,GLP)是药物非临床研究的质量管理和保证体系[2]。美国食品和药物管理局(FDA)于1979年实施了美国联邦法规21CFR58《GLP规范(草案)》[3],主要适用于FDA产品研究或市场许可而进行的非临床研究,其产品主要包括人用药和动物用药、生物制品、人用医疗器械、电子产品、食品和颜色添加剂以及饲料添加剂。我国《药物非临床研究质量管理规范》于2006年正式实施,2017年进行了修订[4]。
FDA要求开展安全性评价研究的非临床实验室符合GLP要求,保证安全性数据的质量和可靠性。FDA的GLP检查包括监督检查和定向检查,监督检查包括定期检查和常规性检查,定向检查包括注册核查、有因检查、跟踪检查和整改检查[5]。GLP常规检查每2年产生一次计划,其他类型检查按照需要安排时间。FDA警告信是FDA针对现场检查中发现的显著违规行为对检查对象发出的警戒,同时在FDA网站上对外公布,是监管过程中主要监管措施[6]。本研究针对FDA在现场检查中针对研究者发布的警告信进行分析,对涉及的法规及条款进行归纳整理分析,以期为我国药物非临床安全性评价研究提供参考。
资料与方法
数据来源
选择2008年至2022年FDA网站上针对非临床研究发出的警告信作为分析数据来源。
分析方法
统计警告信的数量及提出问题的数量,对问题进行分类整理。将警告信提出的问题分别对应21CFR食品和药品条款和我国GLP法规进行分类,描述性统计各类问题。
结果
2008年至2022年期间,FDA对非临床研究发布的12封警告信[7]中共有67项问题数,涉及21CFR58条款120项次,见表1。
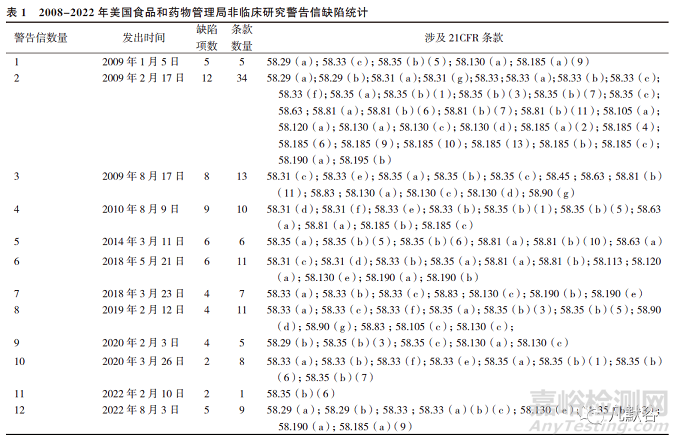
按照21CFR58的分类,警告信中发生率最高的缺陷是质量保证人员、专题负责人、非临床研究方案的实施和非临床研究结果的报告,分别占比21.7%、19.2%、10.8%和10.0%,主要表现在专题负责人及质量保证人员未履行相应的职责,研究未按照方案进行,报告中对数据进行的转换与计算缺少描述等,见表2。
讨论
对2008年至2022年FDA发出警告信进行分析统计,发出的警告信数量及提出问题的数量呈整体下降趋势,表明非临床实验质量在逐年上升。2008年至2018年期间FDA发出的警告信中多次出现标准操作规程不完善不合理的问题,而在2019年及以后未再出现。质量保证人员履职不足、专题负责人未确保研究按照方案进行的问题长期存在,是影响非临床实验质量的重要因素。
2021年FDA未发出GLP警告信,根据FDA2021年GLP现场检查[8]数据统计,现场检查发现缺陷主要是仪器设备管理、培训和专题负责人三个方面,涉及最多的条款是21CFR58.130(a),主要体现在未确保仪器设备使用人员掌握仪器所具备的功能;未确保设施所在工作环境条件是否可满足需要;未确保设施是否运转正常,是否存在会对研究结果造成干扰的因素;仪器使用的培训未能覆盖所有仪器使用人员;专题负责人未能确保研究按照方案进行;实验方案变更未能及时进行记录;未能及时、准确、完整地记录原始数据等。警告信所提问题与FDA在核查过程中发现问题的总体情况可能存在一定差异。警告信中所提的问题仅是针对显著违规行为,若违规者不及时、不充分依据警告信的要求纠正其违规行为,可能会导致FDA向法院提起执行诉讼。因此,对警告信内容进行系统分析,可针对性地对监管模式进行优化,实现更有效的监管,保证公共用药安全。对于一般性的违规行为,FDA通常会向违规者发出无标题信,并在FDA网站上公布,但不公布违规者的相关信息[9]。
GLP认证[10]是指国家药品监督管理局依照申请组织对药物非临床安全性评价研究机构进行GLP情况的检查、评定的过程。与FDA基于风险,针对项目的检查不同,我国重点是对实验机构进行检查。机构只有通过认证检查后,才可以开展相关研究。2022年,国家药品监督管理局食品药品审核查验中心(CFDI)发布的《2021年度药品检查工作报告》[11]显示,2021年GLP认证检查任务38个,不通过的任务1个。GLP认证检查发现的主要问题包括:质量保证部门履职能力不足;机构负责人资质不符合要求;实验动物设施设计与管理不到位;室验环节未能严格执行操作规程;计算机账户权限分级设置不合理等。国家药品监督管理局于2023年1月19日发布《药物非临床研究质量管理规范认证管理办法》[12],进一步规范GLP认证和监督管理工作。
本研究归纳分析2008年至2022年FDA针对GLP警告信,发现质量保证人员履职不足发生频次最高,占比21.7%,这同样是CFDI在2021年GLP检查中发现的主要问题。质量保证部门是GLP体系中重要的一环,通过质量保证人员的检查,确保整个实验仪器设施、人员资质及培训、实验方法、操作、记录等均严格遵守GLP规范。FDA与我国法规均对质量保证部门提出对非临床研究进行检查并将检查结果汇报给专题负责人的要求。质量保证部门应当确保足够的时间间隔检查每项非临床研究,并检查每次实验的书面记录和正确签名记录,确保实验结果的可靠、准确、完整、可追溯。质量保证部门的检查贯穿于整个实验,从方案的审查、实验的进行、报告审核,还有人员、仪器、标准操作规程(SOP)、原始记录等,若对每一项的所有环节与内容进行检查,可能会由于时间或环境条件的限制,无法进行深入的检查,缺乏对高风险点的关注。各实验机构应当根据自身的情况,建立具有自身特点的检查方式,并根据情况变化做出及时调整。FDA在2022年给Toxikon Corporation/ Labcorp Bedford LLC的警告信[13]中指出,质量保证部门未能审核研究报告,无法确保该报告准确描述了实验方法,无法确保实验结果准确反映出实验的原始数据。实验重要指标数据血浆体积计算错误产生计算误差,导致随后的肾血浆流量、肾血流量和肾小球滤过率均产生计算误差,且未能纠正1h时间点时尿量计算问题。2021年10月6日质量保证部门审查后,并未发现此问题。2021年10月14日的修订报告未能纠正所有偏差问题。
警告信中提出报告中缺少使用的基本计算和公式。Toxikon Corporation/Labcorp Bedford LLC与FDA均无法保证最终研究报告中的内容是准确的,质量保证工作的失职使整个研究的有效性受到质疑。警告信中出现频率第二的问题是专题负责人,同时也是2021年FDA现场检查总结报告中的高发问题,此处的专题负责人与我国的专题负责人职责不完全相同。FDA的专题负责人除了对研究的执行负责,还要审查批准实验方案及总结报告。而我国的实验方案及总结方案由机构负责人批准,专题负责人在研究中应当确保研究遵循研究方案,确保所有实验数据均准确记录并进行验证,确保在研究期间或研究结束时将原始数据、文件、方案、样本和最终报告存档,确保遵守所有GLP规定。FDA在2020年给University of Kentucky的警告信[14]中指出其未能遵守研究方案进行研究,无法确保数据的质量和可靠性。此外,未能保留和记录原始数据和样本会妨碍对研究结果的审查、分析和验证。确保完整和准确的研究数据,FDA才能在临床试验开始之前充分评估该研究的总体安全性和风险。专题负责人除了需要有胜任资格,还应加强对研究工作人员的培训与考核。2021年FDA发出的检查报告中也频繁提及:所有研究工作人员都应在研究开始前进行GLP培训,规避相关问题的发生。
非临床研究方案的实施和非临床研究结果的报告也是FDA警告信中提及较多的问题。FDA在2022年给Valley Biosystems的警告信[15]中提出所有实验数据,包括测试系统意外响应的观察结果,均需要准确记录并验证;出现可能会影响非临床研究质量的情况时应谨慎对待,采取纠正措施并记录在案;修订相关SOP,防止此类偏差在未来GLP研究中再次出现,并需提交新的或修订的标准操作规程的相关培训文件。
综上,FDA对于非临床研究警告信中提及频率最高的问题集中于质量保证部门、专题负责人的履职尽责和非临床研究方案的实施及报告。一个可靠的质量保证部门是GLP研究成功不可或缺的,最终报告中细微的误差可能会对结果产生巨大影响;专题负责人的规范操作直接影响到研究的质量;实验过程应严格按照方案实施。GLP体系的建立,有助于把控研究的每一个环节,降低误差,确保结果的真实与准确。本研究提示,监管机构应在落实责任、规范操作、加强培训几个方面重点关注专题负责人及质量保证部门,确保实验数据的真实可靠,避免发生严重违规行为,促进非临床研究质量的提高。




