前情简述
近年来,有一个名词频频出现在人们视野——PM2.5(英文名称:FineParticular matter),系指环境空气中空气动力学直径小于或等于2.5微米的颗粒物。由于其较高的表面活性使其表面容易附积更小的有害物质与此同时较小的粒径可以使其长期悬浮于空气当中。自然界以及人类的一系列活动都会产生PM2.5颗粒,我国的一些区域深受其影响,给当地人的生活质量带来严重的挑战。当其在空气中的浓度越高时所代表的空气质量就会越差,为何会有如此论断,系基于对人体呼吸系统的理解发现,当细颗粒的空气动力学粒径小于2.5微米时这类细颗粒容易伴随着人的吸气过程进入肺内进而沉积在肺部引发一系列病变。下文则是将上述PM2.5更换为药物颗粒,从设计目的、体内吸收过程、产品开发重点以及国内外法规等方面来简要阐述以肺部为主要吸收环境的药物递送系统。
干粉吸入剂(DryPowder Inhaler有名吸入粉雾剂,是将一种或多种微粉化药物与载体组成粉体混合物灌装储存于胶囊或泡囊当中,经特殊给药装置处置之后伴随着吸气过程产生的气溶胶实现药物的肺内沉积进而发挥药物治疗作用的一类新型制剂;干粉吸入剂设计初期主要应用于哮喘、慢性肺阻塞、肺部感染等肺部疾病的靶向治疗,经临床使用价值的不断肯定该技术已经发展成为以肺部为给药环境实现全身治疗作用的药物传递系统。
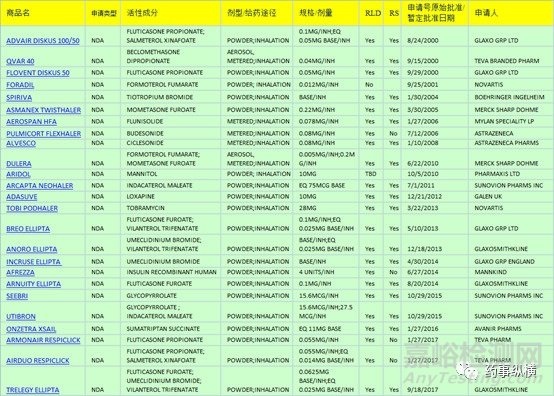
下图即为近年来FDA批准的部分干粉吸入制剂。
基础知识——呼吸系统
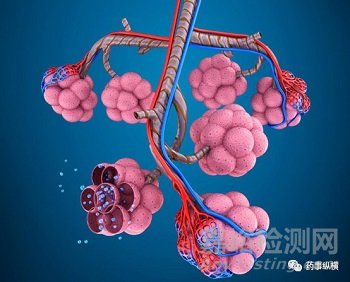
DPI是基于口腔吸入的给药剂型,只有从呼吸的生理机理上理解药物的吸入过程才能实现对产品的深刻理解。人体呼吸系统如下(节选气管到肺泡部分):
呼吸过程简述:吸气时,伴随着的胸腔张肺内压强减小,基于此压差空气被吸入肺部,对于DPI而言在此压差下形成气溶胶实现药物的肺内沉积(一般情况下,该压差大约为4kPa);呼气时,胸腔收缩、肺内压强增大,大于外部压强进而使肺内气体排出,对于空气动力学粒径较小的颗粒在吸气过程中不易沉降在肺内故而伴随着呼气过程外出体外。
基于对呼吸过程的理解以及成像技术的支持,确定了空气动力学粒径与体内沉降部位的一系列关系。
|
呼吸系统 |
沉降粒度范围 |
|
咽喉部位及其以上 |
约≥5μm |
|
气管(主支气管) |
约3~5μm |
|
次级支气管 |
约2~3μm |
|
支气管末端 |
约1~2μm |
|
肺泡 |
约0.5~1μm |
空气动力学粒径小于0.5μm的细颗粒一般会伴随着呼气过程排出。
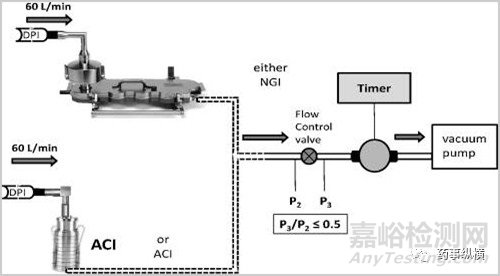
基于对上述过程的理解,开发出了ACI、NGI、MSLI等一系列模拟体内DPI沉降的仪器。
该类设备的工作原理简述为在一定流速以及压差条件下气溶胶在不同孔径的级段内发生撞击并产生沉降进而区分出不同的空气动力学粒度颗粒进而关联体内吸收部位的沉降;基于设备的设计机理只能近似的模拟体内的吸入过程(例如制剂体内的吸入过程是一个在一定潮气环境中的压差递减的吸入过程而上述设备常提供一个均衡的压差);然而对于仿制制剂的开发而言该类设备提供了非常好的体外评价基础。
DPI开发
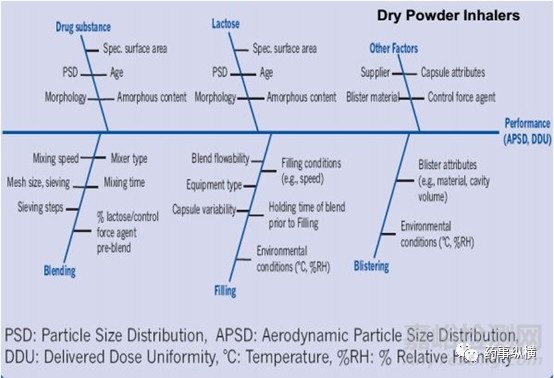
基于对上述体内吸入过程的理解,吸入制剂的开发的重中之重则为如何实现适量药物在肺内的沉积进而实现治疗作用,而对于吸入仿制制剂的开发而言实现药物在呼吸系统各部位的等量沉积是保证治疗等效的重要基础。显然,吸入制剂的关键质量属性除了常规制剂含量、有关物质等之外还包括APSD(AerodynamicParticle size Distribution)以及DDU(Delivered DoseUniformity)等其特有的质量属性。吸入制剂终产品的获得是一系列开发项目综合作用的结果。主要影响因素包括配方、生产工艺、吸入装置以及包装密封系统,具体研究细节方面问题希望同行多多交流。
吸入制剂的开发涉及产品的多个维度,然最核心的维度此处笔者可简述为:吸入制剂的开发的核心是对药物分散、再吸附、再分散的过程理解;是对一系列力学平衡的过程的理解(主要包括原料与原料之间的内聚力以及原料与载体之间的粘附力的平衡,基于此平衡的达成会引入静电力、范德华力、表面张力以及毛细管力等影响因素,故而任何配方开发、工艺开发、装置开发以及包装系统的开发都需以此为指导)。
本文多次提到空气动力学粒度这一概念,众所周知此概念并非指激光衍射法或显微镜方法测定的物理尺寸,实质上该粒度与物理尺寸和物理密度两者综合相关,故而在实际研发当中出现D90内控需在很小粒度之下时切勿大惊小怪。
国内外法规对比
我国15版药典较10版药典在吸入制剂的相关检测方面有了较大的提高,然而与EP以及USP在相关项目的检测方法和可接受标准上依然存在一定差异。主要对比如下:
中国药典

欧洲药典
美国药典
生物等效性研究的要求
我国尚未颁布关于吸入制剂生物等效性方面的评价方法,下文简要叙述EMA以及FDA对吸入制剂生物等效性方面的主要要求细则。EMA和FDA关于吸入制剂生物等效性研究方面各自持有不同的审评理念,这种差异主要体现为EMA认为体外评价的灵敏度高于体内评价故而当仿制产品达到体外APSD等的一致即可满足等效性要求,FDA依然本着最科学的审评态度,生物等效必须建立在生物等效的研究之上。
EMA体外一致即可满足生物等效性的具体要求:
1. 药学一致(剂型、剂量、非活性组分)
2. 目标递送剂量一致(T/R,±15%)
3. 对于DPI装置吸入气流阻力一致(T/R,±15%)
4. 吸入足够量体积时确保活性物质肺内沉积量一致(T/R,±15%)
5. 每组或每级中的APSD分布一致(T/R,±15%)
若上述等效性不能建立则可进行肺内沉积相似性研究,沉积相似则等效性建立;若肺内沉积相似性不能建立则可进行临床以及药效学相似性研究,若建立则相似,若不能建立则“放弃治疗”。
FDA生物等效性要求:
要求仿制药需要证明与参比制剂具有相似的有效性和安全性,包括:装置等效、体外等效、PK等效以及PD/TE等效。
笔者杂谈
如本文图一所示,近年来FDA所批准DPIs大多是基于NDA进行批准,对于该类制剂的仿制开发已然成了众多药企的掘金之处。吸入制剂由于其较低的剂量以及对生产环境的高要求使得该类制剂开发具有较高的硬件要求,国内已有众多企业在该剂型仿制领域内取得了一定的成绩,笔者看来在该类剂型的仿制药开发方面国内依然处于摸石头过河阶段,对于以创新型制剂开发的企业而言吸入制剂或可是一个较好的选择。
参考资料
1.Regulatory Perspetives on Implementing QbD for MDIs and DPIs
2.Guidance for Industry Metered Dose Inhaler (MDI) and Dry Powder Inhaler (DPI) Drug Product document
3.CPMP points to consider on the requirements for clinical Documentation for orally Inhaled Product(OIP) CPMP/EWP/4151/00