摘 要 / Abstract
本文梳理了美国FDA“万络撤市事件”的时间线,通过回顾万络不良反应证据在不同阶段的累积过程,并利用Meta 数据挖掘手段在相应阶段进行动态分析,评估如何及发现万络存在严重心肌梗死风险, 进一步剖析美国FDA 在“万络撤市事件”中监管滞后的原因(包括信息采集来源单一、信号挖掘与分析能力不足、风险评估与决策过程中沟通不畅等)。本文通过总结“万络撤市事件”发生后美国FDA 药品安全监管体系的变革和具体应对举措(包括颁布新的法案、重组机构、开展监管科学研究等),以期从法律体系、组织体系、技术体系等方面进一步为我国药品上市后安全性监测模式的完善提供参考和建议。
This paper first reviewed the timeline of the “Vioxx Withdrawal Event” of the FDA. By reviewing the accumulation of adverse reaction evidence for Vioxx at different stages and using meta-data mining to conduct dynamic analysis at corresponding stages to find out when Vioxx can be found to have a serious risk of myocardial infarction, this paper then analyzed the reasons for the FDA's lag in the regulation of Vioxx, including: single source of information collection, lack ofsignal mining and analysis ability, poor communication mechanism in risk assessment and decision-making. Finally, this paper summarized the changes in the FDA's drug safety regulatory system and specific countermeasures after the “Vioxx withdrawal event”, including the promulgation of new legislation, reorganization of institutions, reconduction of regulatory scientific research, etc., to provide references and suggestions for the improvement of post-market drug safety supervision in China from the legal system, organizational system and technical system perspectives.
关 键 词 / Key words:万络;美国FDA ;药品安全性监测;启示
Vioxx; the United States Food and Drug Administration (FDA); drug safetymonitoring; enlightenment
20世纪90年代,传统的非甾体抗炎药(nonsteroidal anti-inflammatory drugs,NSAIDs),如阿司匹林、双氯芬酸、布洛芬等是当时一线的解热镇痛药物。这些药物在发挥良好消炎、镇痛、退热作用的同时,也可能会造成较大的胃肠道不良反应。机制研究表明,这类药物对环氧合酶(COX)抑制的选择性不佳,在抑制COX-2(发挥解热镇痛作用)的同时也抑制了COX-1(引发消化道出血等不良反应)。
此后,默克公司研发了具有选择性的COX-2抑制剂类非甾体抗炎药罗非昔布片[商品名为万络(Vioxx)],于1999年5月获得美国食品药品监督管理局(Food and Drug Administration,FDA)批准上市,用于治疗骨关节炎、类风湿关节炎,亦用于治疗急性疼痛、原发性痛经。万络自上市后逐渐成为应用最广泛的处方药之一,截至2004年,全球服用过该药品的患者多达8400万人,仅2003年一年的销售额就达到25亿美元[1]。然而,在万络上市后,陆续有文献报道其可能导致严重的心血管不良反应。2004年9月,默克公司自愿召回万络。在万络撤市的过程中,大量相关诉讼案件暴发,引起了舆论的广泛关注。美国FDA及其他药品审批机构受到了诸多知名医学杂志及媒体的指责,称其忽略了万络在安全性方面的早期风险信号,未能主动尽到保护公众安全的职责,导致大量患者陷于本可避免的风险之中。美国FDA在此次大规模药害事件之后进行了深刻反思并持续改革[2]。本文将梳理“万络撤市事件”的发生始末,汇总该事件发生后美国FDA在药品安全监管体系方面进行的改革措施,以期为我国正处于发展期的药物警戒体系建设提供经验借鉴。
1、“万络撤市事件”概述
1.1 “万络撤市事件”始末
万络在上市前总共进行了60个临床试验,共有近5000名受试者参加。基于已有证据,美国FDA认为默克公司已经提供了足够的信息来证明万络的安全有效性[3],于1999年5月批准万络上市,用于治疗急性疼痛、骨关节炎、原发性痛经等。万络上市前安全信息汇总表明,服用该药品的患者很少发生严重的心血管不良事件(发生率小于0.1%)。
为了促进市场推广,默克公司于1999年启动了5个针对万络的大型上市后安全性验证研究,其中万络胃肠道安全性研究(Vioxx Gastrointestinal Outcomes Research,VIGOR)率先完成。VIGOR试验招募了8076名至少50岁(或至少40岁并接受长期糖皮质激素治疗)的类风湿关节炎患者[4],其中4047人被分配接受每天50mg的罗非昔布,4029人被分配接受每天1000mg的萘普生(非甾体抗炎药),主要终点是确认临床上的上消化道事件(包括胃十二指肠蠕动或梗阻、上消化道出血和有症状的胃十二指肠溃疡)。
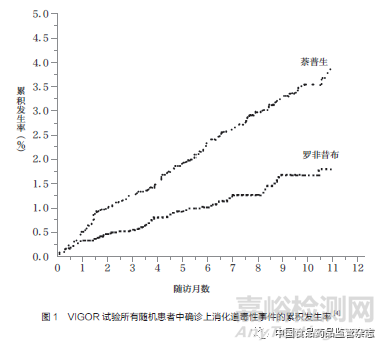
2000年5月,VIGOR试验结果在《新英格兰医学杂志》(The New England Journal of Medicine,NEJM)上发表。数据显示,与萘普生相比,万络的上消化道毒性发生率明显降低[4](图1);在9个月的中位随访期间,使用罗非昔布的每100个患者发生确诊胃肠道事件2.1起/年,而使用萘普生的每100个患者发生4.5起/年。此外,两组的总死亡率和心血管原因死亡率相似。但之后发现此试验结果漏报了3例心脏病发作的严重不良事件(serious adverse events,SAEs)和许多其他类型的心血管不良事件的数据[5]。同年10月,根据VIGOR试验全部SAEs被纳入后的统计数据显示,与萘普生组相比,罗非昔布组心肌梗死等严重心血管事件的发病率较高,为0.5%,而萘普生组仅为0.1%。另外,罗非昔布组卒中、静脉血栓形成和高血压发病风险也显著增多,因此罗非昔布的安全性受到了质疑。默克公司最终将罗非昔布与萘普生引发心血管事件的风险差异解释为:萘普生可抑制血栓素的产生和血小板聚集,具有额外的心脏保护作用,而不是因为罗非昔布的心脏毒性作用。
2001年2月,美国FDA就VIGOR试验结果召开顾问委员会,公布了万络全部的SAEs。委员会认为,罗非昔布的利大于弊,建议修改说明书:不推荐大剂量(50mg/天)长期应用,同时批准罗非昔布新的适应症——类风湿关节炎(25mg/天)。2002年4月,美国FDA责令默克公司修改说明书,增加“可能引发高血压”和“其他心脏疾病”的黑框警告。2002~2004年,许多流行病学研究表明,万络导致患心血管问题的风险增加[6]。此后,默克公司在一项名为“万络预防腺性息肉瘤(Adenomatous Polyp Prevention on Vioxx,APPROVe)研究”的项目中发现,服用万络18个月以上可能会导致患心脏病和中风几率升高。2004年8月,美国FDA在第20届药物流行病学和治疗风险处理国际会议上发布,对于服用大剂量万络(>25mg)的患者,心肌梗死和心性猝死的发病风险比未使用该药的患者高出3倍[7]。2004年9月,默克公司决定自愿召回万络。
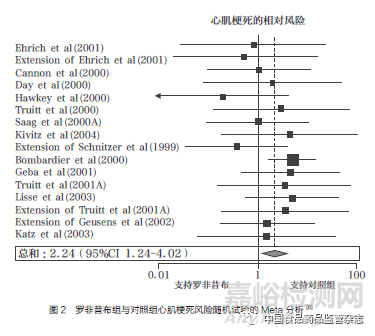
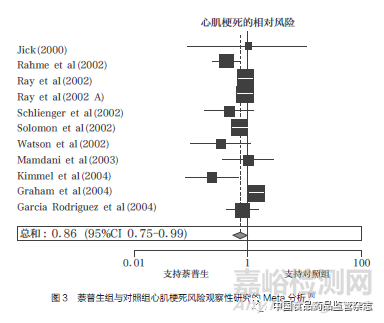
2004年12月,《柳叶刀》杂志(The Lancet)发表了万络导致的心血管事件累积分析,将18项随机对照试验和11项观察性研究的数据结果进行综合分析,证实了罗非昔布会引发心血管不良反应,且使用罗非昔布患心肌梗死的风险是对照组的2.24倍(图2);同时,萘普生的8项病例对照研究和3项回顾性队列研究的数据汇总分析结果显示,使用萘普生患心肌梗死的风险是对照组的0.86倍,仅有微弱的心脏保护作用(图3),且当分析基于与非萘普生的NSAIDs的比较时,也得到了几乎相同的结果,这无法解释默克公司此前所宣称的VIGOR试验中罗非昔布与萘普生相比心血管风险数据异常的原因[8]。
1.2 “万络撤市事件”暴露出的问题
默克公司原本宣称,万络上市后报道的不良反应属于回顾性数据,而非随机双盲试验产生的确认性数据,无法证明万络对心血管系统造成风险。直至其迫于压力,补做了纳入2600名受试者的随机双盲临床试验,才“证实”万络会引发心肌梗死等不良反应。默克公司从未公开披露过能证明万络存在心血管系统风险的临床试验结果,其在2004年9月宣布停售万络后发表的有关该药品安全记录的声明中也承认了临床试验数据披露不完整的行为[6]。
1999年,美国FDA基于默克公司提交的数据批准了万络的上市,但是这些数据直至该药品上市1年半以后才在同行评审刊物上公开发表。在万络的安全隐患暴露后,美国FDA未能及早关注,行使权力强制要求默克公司修改药品标签并督促默克公司对万络的安全性开展重新试验。直到2001年2月,美国FDA关节炎咨询委员会才组织探讨有关万络潜在的引发心血管事件的问题。2004年12月,《柳叶刀》杂志发布的万络心血管不良反应数据的累积分析表明,无论是在长时间还是短时间用药的患者群体中,万络引起的心血管风险都是较大的,且从2000年起,万络引发心肌梗死的风险明显增加[8-9]。如果美国FDA能尽早关注并注意在任何累积综合分析中都能迅速增加与潜在危害有关的新数据,万络就能被更早地采取控制举措,如在万络说明书上添加黑框警告或者将其撤市。在“万络撤市事件”中,美国FDA不仅没有适时将重要的安全数据提供给有关研究人员和广大公众进行独立评估[9],也没有在获得受试者层面的数据后立即对其进行累积分析,导致错失了进行及时且适当决策的机会。
2、“万络撤市事件”的反思及根源分析
2.1 信息采集来源单一且存在遗漏
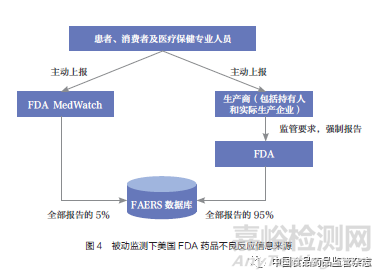
“万络撤市事件”发生时,美国药品不良事件报告系统(FDA Adverse Events Reporting System,FAERS)分别是强制企业报告体系和MedWatch自愿报告体系[又称FDA安全信息与不良事件报告项目(The FDA Safety Information and Adverse Event Reporting Program)],这两大体系是美国FDA获得药品不良反应相关信息的主要来源,其中从生产企业处获得药品不良反应数据的占比约为95%。美国FDA通过外部资源(如来自制药企业、消费者、患者、医疗保健专业人员等多个渠道的报告)收集药品风险信息,这种传统的安全性监测方式被称为“被动监测”[10](图4)。而仅依靠该监测方式的弊端在“万络撤市事件”中暴露无遗:企业出于自身利益考虑,未及时上报失败的临床试验数据;同时,美国FDA由于自身药物警戒能力不足,缺乏主动监测和风险实时预警能力,导致其过于依赖企业递交的风险数据,从而无法及时掌握完整的药物安全性信息来开展流行病学研究。美国FDA既缺乏充分的财力和物力自行开展研究,也缺乏对违规企业进行处罚的法律授权,面对问题束手无策。
2.2 信号挖掘与分析能力不足
由于缺乏安全性信号自动挖掘工具和基于动态数据的风险实时分析技术,美国FDA忽略了万络在安全性方面存在的早期风险信号。其在2000年万络新的中期分析数据公布之后,仍未开展深入的数据研究与风险评估,直到万络上市5年后,才通过第三方机构分析确认了该药品会引发严重心血管不良反应,将其撤出市场。药品上市后安全性研究内容包括评估已知的严重风险信号,发现潜在的非预期严重风险[11]。而检测药品安全信号离不开数据挖掘技术,因此探索建立智能挖掘系统以辅助信号提取工作的开展十分必要。被检测出的信号只是一种潜在的风险提示,还需要对其进一步分析与评价。“万络撤市事件”发生时,美国FDA在数据处理、信号检测、风险评估方面都存在不足。其不仅缺乏相应指南说明如何在观察性数据的基础上科学开展药物流行病学研究,也未建立良好的药物警戒、药物流行病学数据平台,使安全性评价人员能够从提交的不良反应报告中更有效地发现和追踪药品安全信号[12]。
2.3 风险评估与决策过程中沟通不畅
在对万络开展风险评估的过程中,美国FDA新药办公室(office of new drugs,OND)和药品安全办公室(office of drug safety,ODS)存在地位不平等、沟通不到位等问题。ODS作为药品不良反应决策第一责任人,其职权仅限于对药品上市后的审查,而就药品上市前安全问题(包括风险管理问题)仅向OND提供咨询,却没有独立的决策权力。“万络撤市事件”发生时,美国FDA在药品安全方面的决策过程中缺乏清晰的ODS定位,ODS在FDA专家咨询委员会会议中发挥的作用不明确,其安全信息展示和监督权也被有所限制[13]。此外,ODS与OND的审查部门之间缺乏沟通,ODS给出的部分建议未得到重视和遵循。
3、“万络撤市事件”后美国FDA的改革措施
“万络撤市事件”后,美国颁布了新的法规指南,在药品上市后风险管理方面给予了FDA更大的权力和更多的资源,也对相关企业如何做好药品安全性监测工作进行了指导。经过“万络撤市事件”的教训,美国FDA再次审视其工作流程和组织机构,重新界定了药品不良反应及安全性相关监管工作的职能,并根据2007年《食品和药品管理修正法案》(the Food and Drug Administration Amendments Act of 2007,FDAAA)相关规定对药品评价与研究中心(center for drug evaluation and research,CDER)进行改组。
3.1 颁布《食品和药品管理修正法案》
2007年9月,美国颁布FDAAA,通过立法对以下几个方面授予FDA新的权限和资源,从而加强对药品上市后的安全监管[14-15]。
3.1.1 增加药品安全领域经费支持
在财政资源方面,根据《处方药用户收费法》第四版(Prescription Drug User Fee Act Ⅳ,PDUFAⅣ)药品安全五年计划,美国国会批准向FDA拨款2.25亿美元,专门用于FDA中专职负责药品上市后风险管理的监测与流行病学办公室(office of surveillance and epidemiology, OSE),以加强药品安全跟踪监测[11]。PDUFAⅣ作为FDAAA的第一个系列,其财政授权极大帮助了FDA为美国公众开发安全有效的新药,减少了FDA对企业的经济依赖,进一步完善了药品上市后IT系统,开发并验证了风险管理和交流工具。
3.1.2 强制申办方公开临床研究数据
FDAAA明确规定,若申办方要在美国开展药物Ⅱ期及之后的临床试验,必须在美国国立卫生研究院(National Institutes of Health,NIH)下属网站“ClinicalTrials.gov”上注册,并应在试验结束后1年内填报临床试验结果摘要信息,将临床试验结果、严重或频繁的药品不良反应、公共健康建议、药品审批进程、受试者人口统计学及基线特征、统计分析等数据向公众公开[16-17],否则将面临罚款、禁制令和刑事诉讼等惩罚措施。例如,美国马萨诸塞州的制药商Acceleron Pharma Inc.就因未能向“ClinicalTrials.gov”报告临床试验结果而面临了美国FDA发出的首次民事罚款威胁,后又通过发布摘要结果履行了其法定义务。
3.1.3 创新提出REMS概念,更好控制已知风险
FDAAA授予美国FDA权力,可要求申办方制定风险评估和缓解策略(risk evaluation and mitigation strategies, REMS)。通过将REMS写入立法,美国FDA被赋予了对未履行责任企业进行经济处罚的权力,这大大增强了REMS执行效力。为了提升风险评估水平,FDAAA“上市后研究与监测”(Post-marketing Research and Surveillance)章节增加了“505-1REMS”条款。一方面,FDA有权在认为必要的情况下,要求申办方在新药注册时递交REMS评估报告,以确保药品的获益大于风险;另一方面,对已获批的药品而言,如果有新的药品安全信息(如未识别、标注的风险,已知严重不良反应相关新发现),FDA也有权要求申办方提供REMS[11]。
3.1.4 开展主动监测
美国国会在FDAAA中授权了哨点行动项目(Sentinel Initiative),这是一个用以建设和实施国家药品安全电子监测系统的长期项目[18]。2008年5月,美国卫生与人类服务部(Department of Health and Human Services,DHHS)及FDA宣布发起哨点行动。该哨点行动利用各种医疗数据库(如医保数据库、电子病历及药品监管数据库等),将多个数据源整合起来用于药物警戒研究,从而主动监测药品安全[19]。其中,FDA每年购买的商业医保数据库涵盖的数据量大,较完整地收集了参保人员信息,在服药人群安全性信息采集工作的完整性和标准化方面作出巨大贡献。2016年,FDA启动全规模哨点系统,作为药品上市后安全性监测工具库核心资源[20]。哨点系统具有全面实现主动上市后风险识别和分析(active post-market risk identification and analysis,ARIA)能力。FDA通过ARIA系统可实时收集药品全生命周期安全信息,自动开展信号挖掘、验证和确认,能够对特定药品—事件组合进行有针对性的监测,并根据优先等级进行分类,识别和确认新的安全问题。
3.2 发布3个风险管理技术指南
美国FDA为改正自身在药品风险管理中存在的问题,于2005年3月发布了关于药品风险管理的3个最终指南,分别是:《上市前风险评估指南》(Guidelines for Pre-Marketing Risk Assessment),《风险最小化行动计划的开发和使用指南》(Development and Use of Risk Minimization Action Plans),《药物警戒管理规范与药物流行病学评估指南》(Good Pharmacovigilance Practices and Pharmacoepidemiologic Assessment)。这3个指南分别从药品上市前、上市后和企业自身控制3个角度对药品风险管理提出规范性建议[21]。在上市前后的试验和药物警戒方面,FDA吸取了“万络撤市事件”中对于药物流行病学研究不够重视的教训,收录了一系列用于评价药品安全性风险因素的药物流行病学常用方法,并利用登记记录系统和问卷调查等方式多渠道收集信息,并从文献报道、医师记录等来源多方面识别安全信号,评价影响药品不良反应的各个因素。FDA以发布风险管理指南的方式明确企业的风险干预义务,对企业药物警戒义务履行情况的监督以协商沟通为主,辅以强制性干预措施。
3.3调整优化组织机构
3.3.1 成立药品安全监督委员会
美国FDA于2005年2月设立药品安全监督委员会(Drug Safety Oversight Board,DSB)。DSB 由FDA的3个中心以及联邦政府其他8家卫生健康相关部门机构[包括医疗保健研究和质量管理署(Agency for Healthcare Research and Quality,AHRQ)、疾病控制和预防中心(Centers for Disease Control and Prevention,CDC)、医疗保险和医疗补助服务中心(Centers for Medicare and Medicaid Services,CMS)、国防部(Department of Defense,DOD)、卫生资源和服务管理局(Health Resources and Services Administration,HRSA)、印第安人卫生服务署(Indian Health Service,IHS)、国立卫生研究院(National Institutes of Health,NIH)、退伍军人事务部(Department of Veterans Affairs,VA)]的代表组成[22]。DSB 的重要职责之一是帮助FDA评估本部门做出的药品安全决策对其他联邦机构健康卫生体系的影响。根据FDAAA的法律授权,DSB负责在必要时就重要且经常出现的药品安全问题向FDA提出处理和沟通建议,但无权撤销药品或者更改药品标识。此外,DSB还负责为FDA应当何时就药品隐患向消费者发出警告提出建议。
3.3.2 组建新的监测与流行病学一级办公室
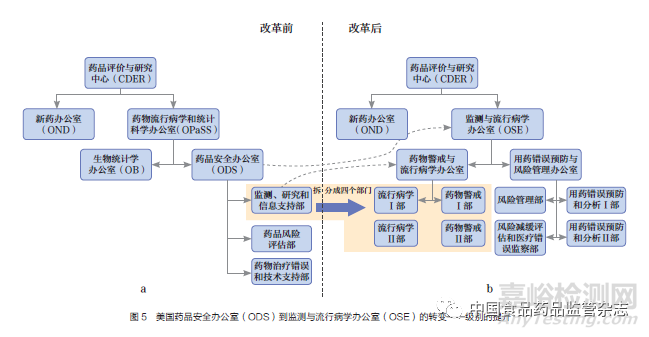
根据FDAAA新授权的职责,美国FDA进行了相应的组织架构调整。组织机构改革前,ODS是FDA负责药品不良反应监测的最主要部门,级别属于二级办公室,隶属于药物流行病学和统计学办公室(office of pharmacoepidemiology and statistical science,OPaSS)(图5a)。组织机构改革后,ODS更名为OSE,并将级别从二级办公室提升至一级办公室,直属CDER的中心主任办公室(图5b)。OSE办公室地位的提升,不仅增强了与其他部门的沟通交流和话语权,还扩大了流行病学、风险评估、项目管理等药品安全相关领域的岗位编制,拥有了更强大的监管力量。机构重组后,OSE主要负责安全信息采集、风险识别和初筛[23](表1),OND主要负责获益及风险整合评估和监管决策,二者协同开展获益及风险监测。

3.3.3 增设药品安全政策和通信人员
2006年4月,美国FDA宣布撤销OPaSS,在CDER中心层面增设药品安全政策和通信人员(safety policy and communication staff,SPCS)岗位,并且任命原OPaSS主任为CDER中心副主任,负责管理SPCS的具体工作。SPCS主要管理DSB和MedWatch,负责药品安全问题和政策勘查,通过FDA网站向医药专家和患者提供药品安全信息[17]。
4、思考与启示
自2015年我国药品审评审批制度改革以来,国产创新药的批准数量大幅度提升。需要注意的是,由于通过大样本量才能有效观测到罕见的、严重的药品不良反应,我国的药物警戒系统应尽早做好准备。美国FDA施行的改革措施使其逐步建立起覆盖全面、职权清晰、协调顺畅的药物警戒体系,这对我国全生命周期药物警戒体系建设和发展有启示意义。
4.1 进一步完善药物警戒法规指南体系
目前,我国药物警戒相关法律法规正处于快速更新时期。自2017年加入国际人用药品注册技术协调会(The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use,ICH)以来,我国相继发布了《国家药品监督管理局关于药品上市许可持有人直接报告不良反应事宜的公告》《药品管理法》《药物警戒质量管理规范》等一系列法律法规和指导文件。建议监管部门进一步完善药物警戒法规指南体系,制定专门的指南性文件,加强对相关企业开展风险管理实践工作的指导。
4.2 进一步完善药物警戒组织体系
在我国目前的药品安全监管体系中,药物警戒涉及的药品上市前审批、上市后监测评价、审核查验等职能分设在不同部门。其中,药品不良反应监测部门的职责侧重于药品上市后的安全性监测评价,药品审评审批部门则侧重于药品上市前的安全性和有效性评价。建议我国结合国情,借鉴美国FDA优化组织机构的做法,调整我国监测机构设置策略,进一步明晰各部门职责和工作程序,建立药品上市前和上市后管理部门之间的信息交流、沟通协作机制,实现上市前和上市后药品风险管理的有效衔接和密切沟通,形成药物警戒不同环节的有机融合[24]。
4.3 进一步健全、强化药品安全信息披露机制
目前,我国《药品管理法》中虽然包含了部分关于药品安全信息披露的内容,但仍存在定义不明确、披露内容较分散、缺乏相应罚则等局限性。此外,部分企业基于自身商业利益的考虑,存在隐瞒其失败试验结果的情形,将患者置于治疗延误或失败的风险之下。因此,建议进一步完善我国药品安全信息强制披露的法律制度,明确规定披露主体的权责,细化针对企业未履行信息披露义务的罚则,进一步完善企业信息披露的奖惩机制,避免在临床试验失败后仍有其他企业重蹈覆辙。
4.4 进一步完善安全性数据集的收集和标准化
现阶段,我国药品不良反应监测工作主要依赖于国家与地方药品评价中心的不良反应监测系统。尽管2016年国家药品不良反应监测哨点联盟(CASSA)的成立,极大改善了我国在药品安全数据收集方面较为被动的局面,但是在数据收集的完整性、标准化、智能化方面仍有较大的提升空间,不同医疗机构和药品生产企业在安全信息收集方面缺乏统一的标准,不良反应报告存在专业术语不规范、信息缺失以及数据可溯源性差等不足[25]。建议拓宽安全信息收集的渠道,加强相关企业对信息收集的主动性和积极性。除了打通医疗机构信息系统的药品安全信息收集渠道外,建议将医保数据库也纳入安全数据集中,构建统一的数据收集模式,并采用标准化的数据模型从中提取数据。
4.5 进一步提升智能化信号挖掘和分析能力
目前,我国药物警戒工作的主要着力点不仅集中在不良反应数据收集,还包括数据的挖掘与分析。随着药物警戒体系的发展,我国逐步建立起以人工和计算机数据挖掘技术相结合的模式,用于开展更准确有效的监测工作。但需要注意的是,近年来国家药品监管部门积累了海量的药品不良反应数据,如何对其高效开展数据挖掘并及时采取风险预测措施,还需要进一步的发展和提升。建议推进数据挖掘和自动预警系统、药物安全性智能评估工具的开发,实现安全性信号的智能化识别,从而实现构建智慧化的全生命周期药物警戒与风险管理体系。
引用本文:周天爱,马倩,杨劲*.“万络撤市事件”后美国FDA药品安全监管体系的变革和具体应对举措分析[J].中国食品药品监管,2023(04):52-73.