单克隆抗体制品(以下简称“单抗”)病毒污染风险控制是一项系统工程,本文梳理了单抗原液生产工艺流程,全生命周期病毒污染风险控制总体策略知识,重点在满足药品生产质量管理规范(2010年修订)和相关厂房设计规范的前提下,针对单抗原液生产的工艺特性,从原液生产工艺流程和除病毒工艺布置方面对单抗原液生产车间的工艺设计进行分析。
1、单抗原液生产工艺流程分析
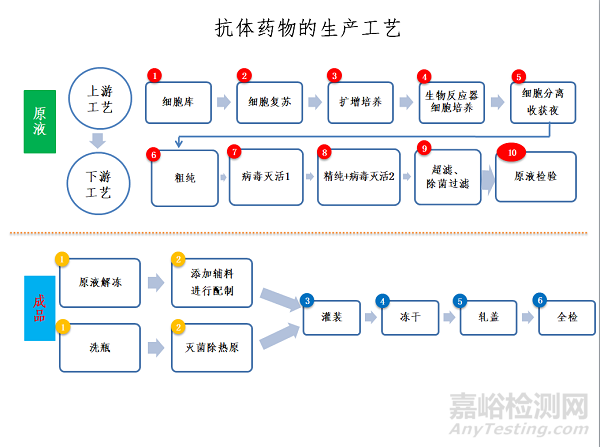
抗体药物的原液生产大体可分为上游工艺(又称细胞培养)、下游工艺(又称纯化)2个环节,每个环节都拥有其核心技术,这些核心技术环环相扣,形成了抗体药物生产企业的核心竞争力。一个典型的抗体药物原液生产工艺流程如下图:
典型的抗体药物原液生产工艺流程
单抗制品下游工艺(又称纯化)的商业化生产流程一般采用以下步骤:粗纯(如亲和层析)→病毒灭活1(如低pH病毒灭活)→精纯(如阳离子层析、阴离子层析、疏水层析)→病毒灭活2(如除病毒膜过滤)→浓缩(如超滤)→除菌过滤→抗体药物原液。
2、单抗全生命周期病毒污染风险控制总体策略
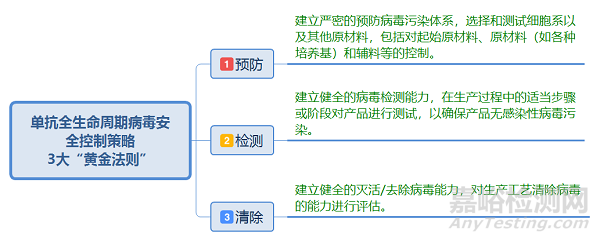
病毒污染可能会对单抗质量和产品供应的潜在影响,可能产生重大后果,生物药企需要谨慎管理病毒污染风险,降低病毒污染的风险,并制定相应风险控制措施,将病毒污染风险控制在最低限度,单抗病毒安全保障需建立整体策略,根据2020《中国药典》三部生物制品病毒安全性控制中提到,“应明确影响病毒清除效果的关键工艺参数及控制范围,并在此基础上建立充分的产品制备工艺过程的控制策略。”,根据ICHQ5A《来源于人或动物细胞系生物技术产品的病毒安全性评价》提炼出从以下三方面入手建立单抗制品全过程病毒安全综合保障策略,即单抗制品全生命周期病毒安全控制策略的3大“黄金法则”:
3、GMP对生物制品的厂房要求
1)法规1—药品生产质量管理规范(2010年修订,2011年3月1日施行)
第四十六条 为降低污染和交叉污染的风险,厂房、生产设施和设备应当根据所生产药品的特性、工艺流程及相应洁净度级别要求合理设计、布局和使用,并符合下列要求:
(一)应当综合考虑药品的特性、工艺和预定用途等因素,确定厂房、生产设施和设备多产品共用的可行性,并有相应评估报告。
(二)生产特殊性质的药品,如生物制品(如卡介苗或其他用活性微生物制备而成的药品),必须采用专用和独立的厂房、生产设施和设备。
(六)药品生产厂房不得用于生产对药品质量有不利影响的非药用产品。
第四十七条 生产区和贮存区应当有足够的空间,确保有序地存放设备、物料、中间产品、待包装产品和成品,避免不同产品或物料的混淆、交叉污染,避免生产或质量控制操作发生遗漏或差错。
第四十八条 应当根据药品品种、生产操作要求及外部环境状况等配置空调净化系统,使生产区有效通风,并有温度、湿度控制和空气净化过滤,保证药品的生产环境符合要求。洁净区与非洁净区之间、不同级别洁净区之间的压差应当不低于10帕斯卡。必要时,相同洁净度级别的不同功能区域(操作间)之间也应当保持适当的压差梯度。
第四十九条 洁净区的内表面(墙壁、地面、天棚)应当平整光滑、无裂缝、接口严密、无颗粒物脱落,避免积尘,便于有效清洁,必要时应当进行消毒。
第五十六条 生产区内可设中间控制区域,但中间控制操作不得给药品带来质量风险。
2)法规2—GMP附录《生物制品》(2020年修订,2020年7月1日施行)
第十三条 生物制品生产环境的空气洁净度级别应当与产品和生产操作相适应,厂房与设施不应对原料、中间体和成品造成污染。
第十四条 生产过程中涉及高危因子的操作,其空气净化系统等设施还应当符合特殊要求。
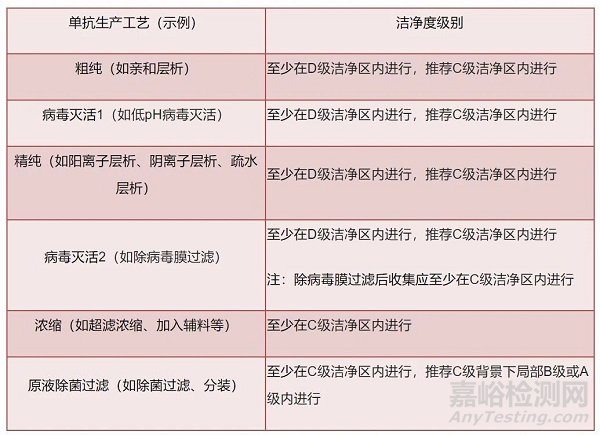
第十五条 生物制品的生产操作应当在符合下表中规定的相应级别的洁净区内进行,未列出的操作可参照下表在适当级别的洁净区内进行:
|
洁净度级别 |
生物制品生产操作示例 |
|
B级背景下的局部A级 |
附录一无菌药品中非最终灭菌产品规定的各工序灌装前不经除菌过滤的制品其配制、合并等 |
|
C级 |
1. 体外免疫诊断试剂的阳性血清的分装、抗原与抗体的分装 |
|
D级 |
1. 原料血浆的合并、组分分离、分装前的巴氏消毒 |
|
2. 口服制剂其发酵培养密闭系统环境(暴露部分需无菌操作) |
|
3. 酶联免疫吸附试剂等体外免疫试剂的配液、分装、干燥、内包装 |
第十七条 灭活疫苗(包括基因重组疫苗)、类毒素和细菌提取物等产品灭活后,可交替使用同一灌装间和灌装、冻干设施。每次分装后,应当采取充分的去污染措施,必要时应当进行灭菌和清洗。
第二十八条 生产过程中被病原体污染的物品和设备应当与未使用的灭菌物品和设备分开,并有明显标志。
3)法规3—GMP附录《血液制品》(2020年修订,2020年10月1日施行)
第十一条 血液制品的生产厂房应当为独立建筑物,不得与其他药品共用,并使用专用的生产设施和设备。
第十四条 原料血浆破袋、合并、分离、提取、分装前的巴氏灭活等工序至少在D级洁净区内进行。
第十五条 血浆融浆区域、组分分离区域以及病毒灭活后生产区域应当彼此分开,生产设备应当专用,各区域应当有独立的空气净化系统。
第十六条 血液制品生产中,应当采取措施防止病毒去除和/或灭活前、后制品的交叉污染。病毒去除和/或灭活后的制品应当使用隔离的专用生产区域与设备,并使用独立的空气净化系统。
4、单抗原液生产工艺布局分析
单抗生产过程中严格执行GMP,以上法规对单抗原液车间生产工艺布局和洁净区环境级别有具体明确的要求,根据目前国内实践经验,单抗下游生产工艺的洁净区域划分原则建议见下表:
另外建议根据所生产产品特性、工艺、预定用途和设备等因素,使用风险评估的手段,采取相应的预防差错、交叉污染、安全防护措施,包括但不限于:
(1)采取必要的措施,防止病毒灭活/去除后的产品被污染;
(2)已经过病毒去除/灭活处理的产品与尚未处理的产品应有明显区分和标识,并应采用适当的方法防止物品混淆;
(3)若使用敞门容器或设备操作时,应有避免污染的措施;
(4)尽量避免同一设备用于不同阶段的纯化操作;
(5)如果使用同一设备,应当采取适当的清洁和消毒措施,并对清洁和消毒的效果进行验证,防止病毒通过设备或环境由前次纯化操作带入后续纯化操作。