DMR、DHF、DHR,这三份档案,最简单的记忆法:做成品的“说明书”、做过程的“履历表”、做合规的“打包证据”
DHF(Design History File,设计历史文件):证明“你是如何把需求一步步变成合规设计”的全过程档案。
DMR(Device Master Record,器械主文档):制造“标准答案”。包含要造什么(产品/软件规格)、怎么造(工艺/设备/环境/检验)、怎么包标配、如何安装维护等。
DHR(Device History Record,器械历史记录):每一批/每一台“按 DMR 造出来”的实绩证明(生产日期、数量、放行、检验、标签、唯一标识等)。
一句话串联:DHF → 输出设计结果;DMR → 吸收最终设计/转产要求,形成制造“蓝本”;DHR → 逐批记录实际生产按DMR 执行。
监管要点
DHF(设计历史文件):21 CFR 820.30(j) 明确要求为每一类器械建立并维护 DHF,含足以证明按设计计划与法规开发的记录与引用。
DMR(器械主记录):21 CFR 820.181 要求 DMR 至少覆盖:产品规格、生产工艺/设备/环境、质量保证程序与接收准则、包装与标签、安装/维护/服务方法。
DHR(器械历史记录):21 CFR 820.184 要求对每个批/台建立 DHR,证明按 DMR 和法规生产,并包含制造日期、产量/放行量、接收与放行记录、标签、设备标识等。
时间线:FDA 于 2024-02-02发布最终规则,将 21 CFR 820 与 ISO 13485:2016 对齐,形成 QMSR;过渡期后(官方宣布的生效安排在规则与 FAQ 中),业界文件术语逐步与 ISO 接轨。
术语变化:QMSR 借鉴 ISO 13485 的 Medical Device File(MDF) 概念。行业评述普遍认为:虽然 DMR/DHF/DHR 的“字眼”在新规文本中弱化/不再出现,但记录实质要求并未降低,可用 MDF 与“设计与开发文件”等对应承接。
对应关系
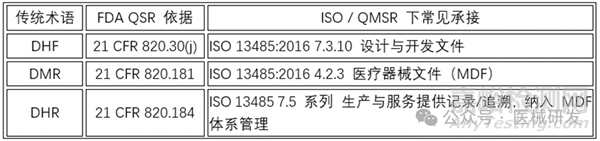
DHF ↔ ISO 13485:2016 7.3.10 设计与开发文件(保存证明符合设计开发要求及变更的记录)。
DMR ↔ ISO 13485:2016 4.2.3 医疗器械文件(MDF)(包含/引用产品全生命周期的程序与规范)。
DHR ↔ ISO 13485:2016 生产与服务提供记录(7.5 系列,含可追溯/标识等)(业界实践将其纳入 MDF 体系下的生产记录模块)。
典型内容清单
DHF 清单(开发侧)
设计计划与里程碑、风险管理主文件的引用、用户需求/设计输入、设计输出、评审记录、验证/确认、临床/性能评估摘要、转产证据、设计变更与配置管理记录等。
ps:设计输入 / 设计输出 的结果,欢迎在留言区留言或在社群中分享
DMR 清单(制造侧)
最终产品或软件规格书、零部件/原材料规范;生产工艺卡、关键工序参数、工装夹具与设备清单/设定值、环境/洁净级别;质量控制计划(进料/过程/成品接收准则、抽样方案、测量设备);标签版式与样张、IFU、内外包设计与方法;安装/维护/服务(如校准/消毒/再处理)程序。
DHR 清单(批次/单台侧)
生产/装配/测试记录(含追溯 ID 与关键参数实测值)、偏差与放行结论、最终标签与 UDI 赋码、返工返修记录。
“落地级”应用示例
举例一次性球囊导管(无源、灭菌)
DHF:用户需求(过载压力/顺应性)、材料选型验证、爆破测试/顺应性曲线、灭菌验证(SAL)、运输验证;
DMR:球囊吹胀参数窗口、热合温度/时间、100% 外观与泄漏测试规范、环氧乙烷灭菌循环、标签与 IFU;
DHR:2025-07-15 批号 B250715;关键工序参数实测表;气密测试全检记录;放行判定与标签样张。
常见问题与实操建议
Q1:DHF 和 DMR 到底谁“归档”谁?
设计移交(Design Transfer)后,DHF 的最终设计输出(图纸、规范、检验标准等)沉入 DMR 成为生产“唯一真相”,后续设计变更要双向联动更新。
ps:虽然说是DHF的最终设计输出,但并不意味着,设计输出的文件是DHF。
Q2:QMSR 时代,要不要把 DMR/DHF/DHR 全改名?
不强制改名,但建议在体系文件中新增“MDF(医疗器械文件)”索引,将 DMR/DHF/DHR 的位置与互联关系以 ISO 13485:2016 的条款映射描述清楚,便于未来审核与多法域合规。
Q3:ISO 13485 下 DHF 有对应要求吗?
有。虽然 ISO 没用“DHF”这个词,但7.3.10“设计与开发文件”的要求与 FDA 820.30(j) 的精神对齐。
速查对照表
QSR→QMSR/ISO 13485
写给工程团队的“最小合规配置”
一个跨文件的索引:建立“产品家族MDF 索引”,把 DHF/DMR/DHR 与相关 SOP、记录表单、风险/临床/变更等关键信息按条款映射收口。
DHF→DMR 的“冻结门槛”:明确哪些设计输出达到“可制造”成熟度才允许写入 DMR,并建立变更后回灌机制。DHR 的“可复核性”:每批/每台的接收准则、关键参数与设备标识、电子签名与追溯要一键导出可审计。