您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-06-05 08:41
摘要
注册检验是药品上市的必要条件,本文旨在不断促进药品注册申请人科学规范地提供申报资料和样品,协助检验机构准确评估药品质量,提升注册检验的质量与效率。 通过梳理《药品注册管理办法》和《药品注册检验工作程序和技术要求规范(试行)》中注册检验的相关规定,针对境外生产化学药品注册检验资料审核工作,结合实践经验,总结注册检验中的共性问题,分析形成原因,并提出应对策略和解决方案。研究发现,注册申请人对境外生产化学药品注册检验工作理解不到位,提供的技术资料、质量标准、样品和标准物质等,存在填报信息不准确、资料不匹配、样品包装材料与申报信息不一致、剩余效期不足等问题。 建议监管机构加强培训指导,加快政策性文件修订,贯通审评与检验的沟通机制;申请人应深入学习理解制度文件,重点关注一致性问题,充分做好检前准备和方法开发,有利于提高申报资料质量,高效完成注册检验。
【关键词】境外生产化学药品;进口药品;注册检验;样品;标准物质;申报资料;共性问题;监管科学
新版《中华人民共和国药品管理法》[1] 和《药品注册管理办法》[2]构建了以人民为中心的药品注册管理体系[3-4],随着我国创新药品上市进程的逐步加快,越来越多的境外生产化学药品在中国获批上市[5-6]。 注册检验是境外生产化学药品在中国境内上市的必要条件,根据审评需要,可由国家药品监督管理局药品审评中心(以下简称“药审中心”)启动或由药品注册申请人(以下简称“申请人”)在审评受理前联系检验机构启动[7],检验工作由中国食品药品检定研究院(以下简称“中检院”)组织口岸药品检验机构实施[2]。 为了促进申请人、中检院和口岸药品检验机构注册检验工作规范化水平的提升,中检院制定发布了《药品注册检验工作程序和技术要求规范( 试行)》2020年版( 以下简称《 工作规范》)[8],但在实际注册检验工作中暴露的一些问题,制约了注册检验工作的高效开展。
本文针对化学药品注册分类中境外生产的1类、2类和5类药品,参照《工作规范》文件要求,结合工作实践经验,梳理总结共性问题,分析形成原因,提出有针对性的应对策略,以期帮助申请人规避常见问题,并为相关制度文件的修订提供参考借鉴,提升注册检验工作质量。 本文针对境外生产化学药品的注册检验(简称为“注册检验”)工作进行研究。
1.境外生产化学药品注册检验工作程序和技术要求
《工作规范》 中附件1.2为化学药品注册检验用资料、样品、标准物质和特殊试验材料的具体要求,是申请人筹备注册检验用材料的重要依据,也是检验机构进行资料审核的参考标准,以下将重点围绕上述要求和申请程序等进行共性问题分析。
2.注册检验共性问题分析
2.1 申报资料的共性问题分析
2.1.1 申请人填报的基本资料
根据注册检验申报的流程,申请人会依次向药审中心提交《注册申请表》、获得检验通知单或补充资料通知、在口岸药品监督管理局获得进口通关单、在中检院进口药品注册检验平台填报打印《境外生产药品注册检验登记证明网上确认表》(以下简称《网上确认表》)[9],其中涉及药品名称、药品生产厂家、包装规格、批号等易出现问题的信息[10-11],对应信息的一致性关系详见表1。 需要注意的是《注册申请表》作为药品注册工作的起始材料,其填报信息将作为后续工作的重要参考,应给予高度关注,同时《网上确认表》 作为注册检验环节申请人信息自查和中检院资料审核的重要文件,同样应予以重视。
▲表1- 注册检验用资料信息一致性对应表
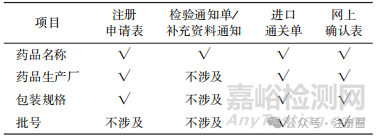
药品名称、药品生产厂、包装规格的信息在国家药品监督管理局(以下简称“国家药监局”)网上办事大厅药品业务应用系统、中国国际贸易单一窗口、中检院进口药品注册检验平台均需申请人分别填报,因此应完全相同,即上述机构要求的《注册申请表》、进口通关单和《网上确认表》中相关信息应一致。 在工作中发现存在资料中药品生产厂不一致的情况,其主要原因为申请人在不同平台填报时使用的生产厂家简称不同。 批号信息仅需在药品通关和注册检验时填报,因此进口通关单和网上确认表中的批号信息应一致,造成批号不一致的情况,多为申请人粗心填错所致,如批号编制较复杂的原料药,容易造成漏填部分信息或将字母“O”错写成数字“0”等。
2.1.2 质量标准和药品通用技术资料
申请人申报的质量标准是检验机构开展注册检验工作的唯一标准,药品通用技术资料反映了质量标准检验开发的完整性、方法的可行性和质控指标的合理性。《工作规范》要求申请人提供按照现行版《中华人民共和国药典》 格式整理的质量标准和起草说明,未强制要求检验方法必须采用《中华人民共和国药典》收载的方法。
对于境外生产药品,通常采用全球化的运营模式,使用全球统一的放行标准,检验方法多参考原研国家的药典开发,其中存在的问题多集中在申报的质量标准注明按照《中华人民共和国药典》制定,而方法验证等药学资料依据国外药典研究,例如:各国药典澄清度检查法中浊度标准液的浊度级号、溶液的颜色检查中标准比色液色号、重金属检查操作中酸的选择、溶出度判定方法的不一致,以及含量均匀度的操作方法和判定标准不一致,使得质量标准与方法验证材料不匹配,不同的检验方法以及不同的判定标准,造成了检验机构无法按照申报的质量标准报告检验结果[12-13]。 在实际工作中发现微生物相关检验项目此类问题也比较突出。 涉及微生物检验的项目主要为注射剂和无菌原料等无菌产品的无菌检查,以及非无菌制剂和原料药的微生物限度检查,申请人提供的质量标准中微生物相关项目参考《中华人民共和国药典》 四部1101无菌检查法制定,其对应使用的应为中国医学微生物菌种保藏管理中心(CMCC)供应的菌株,而方法验证参考《美国药典》等国外药典开发,对应的应为美国典型菌种保藏中心(ATCC)供应的菌株,但目前《中华人民共和国药典》和国外药典就该问题还未协调一致,微生物检验用标准菌株尚未互认或对等收载[14],无法对等使用相应菌种开展检验。 另一类造成不一致的原因多为翻译错误所致。 如国外厂商提供的外文材料要求使用磷酸二氢钠配制缓冲溶液作为溶出介质,而中文的质量标准错误地翻译为了磷酸二氢钾。又如将外文中磷酸盐缓冲溶液配制方法的称样量由7.1g错误地翻译成了0.71g,导致缓冲液浓度降低10倍,这都极有可能使得检验数据无法真实反映药品的溶出特性。 此外,还存在申报的质量标准有些检项关键参数缺失、检验方法表达不清等现象,都将会影响注册检验的开展。
为顺利开展标准复核工作,中检院在《境外生产药品注册检验资料目录》中设计了“复核项目名称”供申请人填写。 对于申请复核项目的名称,应依据申报的质量标准进行明确,对于发补检验的项目,其名称应参照药审中心补充资料通知中“质量标准”明确的项目名称进行确定,即申请复核项目的名称应与申报质量标准项目名称以及补充资料通知要求的项目名称一致,避免pH值、酸度、酸碱度的混用,以及甲磺酸烷基酯误写为甲磺酸烷基脂等问题的发生。
2.1.3 样品出厂检验报告书
《工作规范》要求申请人提供的送检样品出厂检验报告书应按申报质量标准检验,这样不仅可以证明境外生产厂家对未来在中国上市的药品具备相应的检验能力,保障药品质量,也可方便口岸药品检验机构对复核检验数据进行比对。 但部分申请人提供的出厂检验报告书采用内控方法或放行标准进行检验,所使用方法和限度与在中国申报的质量标准不一致,抑或是存在提供工艺验证批的检验报告书与实际送检样品的批次不符等问题。
2.2 样品的共性问题分析
2.2.1 样品的包装和剩余效期
注册检验工作不仅为了考查检验方法的可行性,为准确评估未来在中国上市药品的质量,对送检的样品包装和剩余效期同样做出了要求。 《化学药品注册受理审查指南(试行)》[15]规定,在中国提交申报注册时需提供境外药品管理机构出具的允许该药品上市销售证明文件,即除1类新药外,药品在国内提出注册检验申请前,应已在境外获批上市,具备商业化规模生产能力,可以提供上市包装样品。 但在实际的工作中发现,存在样品信息与注册申请信息不一致的情况,具体表现为在上市申请或补充申请时,使用白盒样品用于检验,还有在以更换生产厂家为目的提出补充申请时,提供原生产厂家样品的情况。 在原料药的注册检验中,常见的问题是送检样品的直接接触药品包装材料与申报的包装材料不一致,《工作规范》允许在适当的条件下选用与申报相一致的包装材料进行分装后送样,例如:申报的直接接触药品包材为聚乙烯袋,申请人应选择相同材质的聚乙烯袋进行分装,不宜更换成玻璃瓶或塑料瓶分装样品,以保证包装材料对药品的质量不会产生影响。 对于涉及微生物限度或无菌项目的,为了避免样品污染,还应提供用于该检项的独立包装样品。
样品的筹备期间,申请人还需重点关注送检样品的剩余效期。 《工作规范》规定样品的有效期应距有效期末一般不少于2个注册检验周期,对于同时进行样品检验和标准复核的,应当不少于180个工作日,仅进行样品检验的,应当不少于120个工作日。 在实际工作中了解到,申请人通常需从其他国家市场调运药品或要求生产厂家生产新批次样品用于注册检验,调回已发往其他国家销售的药品,其时间往往是不可预计的,而对已经做好排产计划的生产厂家提出新的生产需求,则需要等待更长时间。在实践工作中曾出现申请人提出了泡罩包装药品的注册申请,但实际提供了瓶装样品,待其重新准备好泡罩包装样品,时间已经过了1年之久。
2.2.2 样品的送检量
申请人在准备处方相同的液体制剂、半固体制剂(如软膏、乳膏等)时,如审评中心没有特殊的要求,存在有多种规格的,可按《工作规范》要求,根据具体情况确定一种规格的 3 批样品和其他规格至少一批样品进行检验。 例如:造影剂碘佛醇注射液存在 50 mL: 17.5 g(I),100 mL:35 g(I),50 mL: 16 g(I),100 m:32 g(I)等多种规格,在临床使用时根据造影目的的不同,会选择使用不同的剂量规格[16],由于前后两两规格浓度相同,因此申请人可在前2种包装规格中选择一种送检3批样品,另外一种送检1批,后2种包装规格亦是如此。 此类情况在大输液类品种中较为常见,如准备全部规格各 3 批样品,易对申请人筹备样品造成压力,也会造成检验资源的浪[17]。
2.3 标准物质的共性问题分析
注册检验用标准物质在有关物质和含量测定中发挥了重要的“标尺”作用[18]。 《工作规范》规定申请人应随送检样品提供标准物质的检验报告书及相关研究资料,如果为中检院、美国药典委员会、欧洲药典委员会等官方机构发布的标准物质,可提供检验报告书等材料。 如果为申请人自行标定的,还需提供结构确证、含量测定等研究资料,但若申请人提供的资料信息不全或赋值不准确,将无法准确反映检验结果。
与此同时,由于每种标准物质标定的时间可能不同,存在所有标准物质无法同时满足剩余效期要求的可能,为满足送检要求,申请人可能会分批重新标定,造成互相等待的情况。 标准物质的剩余效期要求与样品相同,但申请人多关注样品而忽略标准物质,造成送检时剩余效期已不满足要求。
2.4 特殊实验材料的共性问题分析
对于超出现行版《中华人民共和国药典》 标准中使用和其他不易获得的特殊实验材料,包括检验所需使用的特殊色谱柱、特殊试剂等,申请人应同时送检并提供必要的使用说明文件。 特别是对于一些已经停产或境内存有量较少的特殊仪器,申请人应提前做好调研沟通,确保检验机构具备相应仪器及配件,并提供特定型号的试剂耗材,评估不同厂家试剂对检验结果的影响,保证检验结果的准确性。
3.应对策略与建议
3.1 加强注册检验培训指导
新版《中华人民共和国药品管理法》和《药品注册管理办法》 发布实施后,国家药监局、中检院、药审中心配套发布了多项文件,对注册检验的工作程序和技术规范都提出了具体要求。 建议监管部门加强工作要求培训讲解,持续收集汇总申请人常见问题并公示解答,指导申请人规范注册检验工作。 申请人自身应及时跟踪学习最新文件,加强业内交流,分享经典案例,掌握工作动态,做好内部培训,提升注册人员专业能力。
3.2 加快《工作规范》等政策文件修订
《工作规范》 是指导申请人规范准备注册检验用资料、样品和标准物质等材料的重要文件,也是中检院执行审核工作的重要依据,其在规范注册检验工作的同时,也为申请人提供了一个公平竞争的平台。 随着创新药品全球同步研发申报等新情况的增多,《工作规范》已无法解决现阶段注册检验工作中出现的新问题、新矛盾。 建议加快《工作规范》的修订工作,充分听取申请人、行业协会、监管部门、审评机构、检验机构等相关单位在执行过程中发现的问题和提出的建议,在坚持“四个最严”基本要求的前提下,以有利于人民群众用药安全和服务药品监管为目的,进一步优化程序[19],探讨从审评环节开始的信息化贯通、多渠道沟通交流以及科学制定送检量等方面的可行性,以适应日益增长的全球同步研发现状,满足人民群众的用药需求。
3.3 充分做好检前准备
关注申报资料、样品和标准物质的一致性问题,提供符合要求的注册检验用材料,是顺利进入注册检验环节的最快方法。 样品和标准物质如存在问题,往往需要较长时间才可解决,考虑到跨国公司全球供货的生产模式,建议申请人规划好注册检验申请的时间节点,提前做好样品排产和标准物质标定工作,结合质量标准准备适宜的标准物质,如仅需进行系统适用性确认,可提供系统适用性溶液或混合标准物质,而需进行定量检验的,则应提供单独组分的标准物质,保证剩余效期符合规定,同时充分考虑危化品等特殊试剂的运输条件和备案要求。 对原料药而言,建议根据品种的特性,选择有资质和条件的实验室进行分装,避免湿度影响或微生物污染,保障样品质量。
3.4 完备检验方法开发
发补检验多为上市许可申请和补充申请审评中,药审中心基于风险提出的质量标准单项复核或部分项目复核[20]。 建议申请人在制定质量标准前,做好充分的调研和质量研究工作,结合申报品种在各国药典中的收载情况、ICH和药审中心发布的指导原则等,统筹考虑《中华人民共和国药典》 的特点,科学、完整、合理地制定质控项目和限度。 在开发检验方法时,还应考虑检验用仪器设备的适用性和耐用性,尽量选取普适性较好的通用型仪器,避免使用过时或停产的仪器。 同时建议申请人关注国际药品研发动态和突发事件,如药品基因毒性杂质事件等[21-23],及时开展产品评估和方法开发,以便能够高效应对品种的补充申请或发补检验。
如因发补需再次启动检验的,建议申请人与药审中心加强沟通,在明确检验项目后向中检院提出注册检验申请,在此期间,申请人应尽快确认已有技术资料能否支撑发补检验开展。 一方面,如需建立新质控项目或开发新检验方法的,应关注发补时限,在规定的时限内完成新方法的开发;另一方面,在开展发补检验时,申请人仍需按照《工作规范》 要求,准备注册检验用资料、样品和标准物质,特别需要注意的是,发补检验用样品的自检报告书,应按发补要求采用新方法出具,而非按照原检验方法和限度要求进行检验。
参考文献
[1]全国人民代表大会常务委员会,中华人民共和国药品管理法[EB/0L].(2019-08-26)[2024-08-16]. http://www.npc.gov.cn/npc/c2/c30834/201908/120190826 300489.html.
[2]国家市场监督管理总局,药品注册管理办法[EB/0L].(2020-01-22)[2024-08-16]. https://www. samr. gov. cn/zw/zixxgk/fdzdgknr/fgs/art/2023/art _3275cb2a929d4c34ac80042162a9c257.html.
[13]李文龙,门瑛璇,魏京京,等.新时代中国境外生产化学药品注册检验的变化、机遇与挑战[J/0L].中国现代应用药学,1-5[2025-05-07].https://doi.org/10.13748/j.cnki.issn1007-7693.20242401.
[14]王平,杨胜,张建武.新时代药品注册管理体系的设计与构建--2020 年版《药品注册管理办法》的新理念、新内容、新要求及实施进展[J,中国食品药品监管,2021.1(6):8-17.
[5]安抚东,推动新时代药品检验事业高质量发展的实践与探索[J]、中国食品药品监管,2024.1(12):8-15.
[6]王淑芳,田丽娟.《药品注册管理办法》(2020)实施前后纳人优先审评程序的药品注册情况分析[J].中国新药杂志,2024,1(33):18-22.
[7]蔺娟,乔利涛,李源,等.现阶段药品注册检验启动工作要求及面临的挑战[J],中国药事,2023.3(37):245-249.
[8]中国食品药品检定研究院,关于发布《药品注册检验工作程序和技术要求规范(试行)》(2020年版)及有关事项的通告[EB/0L].(2020-07-01)[2024-08-16]. https://www.nifde.org.cn//nifde/xxgk/zcfg/1fg/20200701134238784.html
[9]王岩,孙磊.简述我所进口药品注册检验的电子化管理[J]中国药事,2007.1(12):966-967.
[10]陈佳亮,孙勇,吴蓉蓉.进口药品通关攻略[J].中国海关,2022.(10):40-42.
[11]黄哲,曲若暄,兰毅鹏,等.罕见病药品进口流程与监管建议研究[J/OL].沈阳药科大学学报,1-7[2025-04-15].http ://doi.org/10.14066/i.cnki.cn21-1349/r.2023.0295.
[12]王立新,马步芳、张培培,等.《中国药典》四部通则澄清度检查法中可能存在问题的探讨[J].药学与临床研究,2021(3):219-223.
[13]步艳艳,任红敏,王智超,等.国内外药典中重金属检查方法及限量比较分析[J].广东化工,2018.1(10):136-138.
[14]朱冉,许华玉,张军,等,中国药品微生物标准体系的建立与发展[J],中国药品标准,2023,24(6):561-566.
[15]国家药品监督管理局药审中心,关于发布《化学药品注册受理市查指南(试行)》的通告(2020年第10 号)[EB/0L],(2020-07-02)2024-08-16].https://www.cde. org.cnmain/news/viewlnfoCommon6386d7ca5al515db4259acb1 b8f16333.
[16]高慧芳,彭瑛.造影剂脑病的研究进展[J].中国临床神经外科杂志,2021,26(12):964-966.
[17]薛品,祁文娟,黄清泉.降低现行药品检验抽样量的可行性探讨[J,中国药事,2022,36(11):1234-1238.
[18]谭丽媛,蔡形,王岩,化学药品标准物质定量分析方法研究进展[J],中国药物警戒,2023,20(11):1209-1216.
[19]李文龙,王雪蕾,谭丽媛,等.境外生产化学药品注册检验工作机制创新研究[J],药学研究,2025.44(3):296-301.
[20]李源,蔺娟,史利威,等.新形势下我国药品注册检验启动与实施工作要求及挑战[J.中国临床药理学杂志,2023,39(5):757-760.
[21]张帆,吕明,佟飞,等.化学药物中亚硝胺杂质的控制策略及审评考虑[J,中国药事,2023.37(12):1431-1437.
[22]袁松,李捷,张娜,等.UPLC-MS/MS 法测定磷酸西格列汀中基因毒性杂质 NTTP[J].药学研究,2023,42(12):1000-1004.
[23]黄海伟,袁松,张娜,等.UHPLC-MS/MS 法测定盐酸普萘洛尔缓释片中基因毒性杂质N-亚硝基普萘洛尔[J.药学研究,2023,42(7):481-484.

来源:Internet


