您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-05-29 08:32
药用辅料是药品的重要组成部分,辅料可能是药品的主要成分(占比80%~90%),并影响药品的整体性能。虽然辅料通常被认为是没有药理学活性的,但在制剂的制备中不可或缺。例如,辅料可以通过确保活性物质在制剂中的化学和物理稳定性(如抗氧化剂和稳定剂),保护其在储存和使用过程中免受微生物污染(如防腐剂),或优化其生物利用度(如增溶剂、渗透促进剂以及改变药物从剂型中释放的物质)。此外,某些辅料在确保患者依从性方面也发挥着重要作用,例如,矫味剂和甜味剂可确保口服制剂的口感。
在欧盟(EU),辅料被定义为“药品中除活性物质和包装材料之外的任何成分”。这种既宽泛又模糊的定义恰恰反映了这些物质的复杂性和多样性。辅料可以根据其功能(如抗氧化剂、防腐剂和填充剂)、来源(如合成产品与天然产品)、结构组成(如单一化学实体与化学相关的化合物混合物;合成产品与半合成产品)或使用经验(如传统辅料与新型辅料)分为不同的类型。
除了具有悠久使用历史且被广泛接受的药典或制剂手册中描述的传统辅料。更复杂、更新型的辅料,在现代先进药物递送系统和生物技术药物的开发中变得不可或缺,对这些新型辅助性辅料的需求正在迅速增长。
那么什么是新型辅料?根据当前欧盟指南,新型辅料是指首次在药品中使用,或者首次用于新的给药途径的物质。它可能是一个全新的化学实体,也可能是之前未在药品中使用过的已知物质。此外,如果某种药用辅料首次用于特定给药途径,也被视为新的。例如,通常用于口服产品的药用辅料,当首次用于眼部时,也被视为新型辅料。FDA将新型辅料定义为在预期使用场景(如预期暴露水平/持续时间或给药途径)中尚未通过现有安全性数据完全验证的辅料。NMPA则认为“新药用辅料”是指拟添加到治疗用或诊断用药物中的任何无活性成分。根据现有的安全性数据,尚不能充分评估用拟定的剂量水平、暴露持续时间或者给药方式使用时对人体的风险。
现有的辅料非临床评价的监管指导文件(如FDA、EMA、ICH等)虽然有用,但相对过时。FDA关于辅料非临床安全性评估的指导文件发布于2005年。NMPA《新药用辅料非临床安全性评价指导原则》发布于2012年,该指南主要引用的FDA这个版本。目前尚无国际统一的指导文件来明确非临床试验的要求以支持新型辅料的验证。而且,行业在新型辅料开发方面的经验相对有限,可能会带来不确定性及战略风险,也可能会阻碍创新并抑制公司对其的使用/开发。因此,患者可能会被剥夺使用新的、有价值的药物的机会,或者那些可能显著改善老年患者和儿科患者等脆弱群体用药体验的药物。FDA和NMPA关于新型辅料非临床安评的要求大致如下表所示。
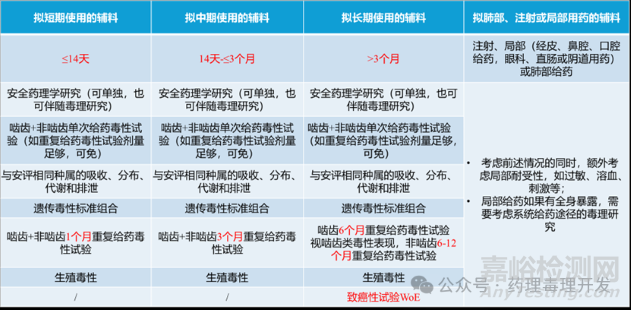
当然,指导原则也提出,对于一些情况,可以不必开展完整组合的毒理学评价。比如一些辅料现有的人体安全性数据可以替代某些非临床安全性数据;以往已经有与拟定用途的暴露环境相关人用资料的辅料;某些情况下(例如相似的给药途径、暴露水平、患者群体和暴露持续时间),以往的使用经验可以充分提示一种辅料的合理性。
指导原则毕竟颁布时间比较久,结合IQ Consortium Novel Excipient Working Group (NEWG)开展的一项制药公司调查,看下工业界具体执行情况。这项调查旨在识别新型辅料非临床评估的主要问题或限制。研究共收到了33个项目的反馈,其中14个项目正在进行中,19个项目已完成。约一半的项目(17个)采用已有数据和新的非临床研究相结合的模式。其余项目仅开展新的研究或仅依赖现有可用数据,各占一半。
有两个项目仅使用现有数据(均用于在更高剂量下辅料的使用),采用了“类比”方法(read-across approach),即利用其他分子的安全性数据作为支持性证据,且均获得了FDA的认可。
33个项目中,9个为全新辅料项目,其中4个项目仅开展新的非临床研究,5个项目结合新的研究和现有数据。24个辅料项目是拟用于新的用途,其中7个项目仅使用现有数据,12个项目结合现有数据和新的非临床研究,5个项目仅开展新的非临床研究。
将项目根据药物形式分为生物药、多肽和小分子化药。
其中反馈的抗体项目5个,ADC项目2个,所有项目均设计新型辅料的长期用药。新型辅料的具体研究方法包括:1个抗体项目和1个ADC项目仅使用现有数据来支持更高剂量或更长期使用。其余项目旨在通过新的毒理学研究(有或没有现有数据)鉴定辅料用于更高剂量或新的给药途径(吸入或眼部)。1个抗体项目和1个ADC项目计划使用现有数据和有限的额外研究来鉴定新型辅料。抗体项目包括大鼠药理学研究和重复给药毒理学研究(大鼠和犬),ADC项目则开展了重复给药毒理学研究、毒代动力学(TK)研究和吸收、分布、代谢和排泄(ADME)研究(均在大鼠中进行),同时还进行了体外(代谢和整体药物毒理学)和计算机模拟(遗传毒理学)研究。除了提及的整体药物毒理学是采用的药物开展研究,其余研究用的受试物均是新型辅料。剩下的另外3个项目在一种或两种种属中开展了安全药理学和重复给药毒理学研究(其中1个项目使用了转基因小鼠)。最后,以上这些项目均未包括致癌性研究。
再看下多肽类药物。共统计了7个多肽产品,其中口服途径5个,其余为皮下或眼部给药。口服给药多肽产品中,2个属于新型辅料,开展的非临床研究涵盖安全药理学、遗传毒性、重复给药毒性、生殖毒性、TK、代谢和致癌性研究(涉及一种或两种种属)。还有2个产品属于提高已批准辅料的剂量,其中一个项目与上述新型辅料项目类似(不包括致癌性研究),另一个项目增加了代谢研究,以评估新型肠溶包衣的影响,并探索观察到的靶器官毒性的原因。最后1个项目属于已有药物和食品使用历史的辅料认证,通过文献综述的方式进行的认证,并获得监管机构认可。辅料增加皮下注射途径的多肽项目,使用了现有数据并开展额外的局部耐受性研究(涉及两种种属)。辅料增加眼部(玻璃体腔内)途径的多肽项目,开展了小型猪玻璃体腔内注射重复给药毒性研究(包含TK)。
涉及小分子药物辅料的项目共17个,分为两类,一类是在长期给药过程中添加的新型辅料,一类是已有辅料的新应用,如新给药途经。
大多数项目采用辅料开展了完整的毒理学研究,包括遗传毒性、安全药理学研究、单次给药和重复给药毒性研究、生殖毒性,以及偶尔的致癌性研究。
对于已有辅料的新应用,如新给药途径,可以开展有限的毒理学研究。包括使用现有数据和少量额外研究,通常仅涉及一个种属。
有些产品仅短期(2个)或中期(2个)使用。这种情况与已有辅料新应用(如新的给药途径和更高剂量)不同。其中3个项目仅使用现有数据,而另一个项目(短期使用)在现有数据的基础上,增加了新的遗传毒性、安全药理学、重复给药毒性、生殖毒性、TK和代谢研究。
最后提一下新方法(NAMs)在辅料安全性评估中的使用。NAMs包括非动物方法,如计算机模拟(如定量构效关系或生理基础模型)或体外(如类器官培养)系统。这一理念最近几天被FDA非临床研究减少动物使用的公告炒的沸沸扬扬。这次调查结果显示,33个辅料项目中,只有2个使用了NAMs。而且,这两个项目的NAM数据是与一系列体内研究数据一起使用,一并提交的。辅料尚且如此,新药更不太可能短期内采用NAMs替代动物。
关于监管机构的审评差异
在33个项目中,12个项目已向美国和/或欧洲卫生当局提交数据;15个已至少向一个其他区域提交数据;6个尚未提交给任何监管机构。其中有3个项目出现了不同监管机构的审评差异。均属于小分子药物中的辅料应用,适应症为肿瘤(1个)或呼吸系统疾病(2个)。
肿瘤药物案例涉及一种辅料混合物,其成分拟用于比其他药物产品更高的剂量,并通过新的给药途径(静脉注射)使用。辅料混合物包括成分A(已在批准产品中作为反离子使用)和成分B(聚合物混合物)。药物产品以纳米颗粒形式配制。监管方面,欧洲和美国均同意有限的非临床方案(仅在两种种属中进行重复给药毒性研究)以支持1期和2期研究。然而,支持3期研究所需的研究内容存在差异。欧洲要求成分A进行两种种属的颗粒分布研究和与已批准药物产品的系统暴露对比,美国则要求进行遗传毒性评估。
吸入药物案例,欧洲和美国对申办方提出的在犬中进行3个月药物产品毒性研究以及不进行致癌性评估的提议给出了不同意见(基于辅料的性质)。FDA接受不进行任何致癌性研究,而欧洲监管机构要求进行一种种属的致癌性研究。欧洲还要求在犬中进行更长周期(六个月)的药物产品毒理研究,而FDA除了在犬中进行3个月的药物产品毒理研究外,还要求进行一项大鼠等效性研究。在后续与日本监管机构的沟通交流中,其认为“辅料”方案存在缺陷,要求进行进一步的慢性毒性(两种种属)、致癌性评估(两种种属)和开展有限的生殖毒性研究。另外一个吸入药物案例,虽然美国和日本卫生机构接受了药企提议的非临床方案,但欧洲机构要求提供额外的辅料代谢、体外次要药理学和安全药理学数据。
可见不同监管机构对于辅料的非临床研究方案要求也不完全一致。
对辅料非临床评估指南的反馈与建议
指导原则改进需求:当被问及目前关于新型辅料非临床评估的指导是否可以改进时,17家公司表示“是”,仅有2家公司表示“否”。同样,当被问及是否需要一个统一的辅料非临床开发指导时,18家公司表示“是”,仅1家公司表示“否”。涉及到的具体反馈与建议包括:1)统一指南的重要性。许多受访者认为,统一的指南(如ICH 指南)对于协调和简化不同国家监管机构的要求非常重要;2)FDA指南的局限性,FDA 2005年发布的指导已经过时,可能无法充分涵盖现代辅料的使用,例如在一些先进的药物递送系统中的应用;3)指南的多样性解释。当前的指南容易被不同地区监管机构解释为需要更多数据,这可能导致企业采取保守策略,开展不必要的额外研究;4)开发成本与时间负担。鉴于辅料开发的高成本和时间负担,建议未来的统一指南应包括针对不同开发阶段所需研究的详细信息,以加快辅料进入临床的速度;5)数据接受标准的明确性。指南应明确哪些类型的数据可以用于辅料资质认证,例如文献数据、类比数据和NAMs;6)药物产品与辅料单独研究的对比。建议增加关于药物产品与辅料单独研究要求的明确性,包括是否需要开展特定的桥接研究;7)现有数据的利用。建议更广泛地获取和利用现有辅料数据以支持其在新场景下的使用,或者使已经由监管机构审查过的数据更易于查阅、获取;8)独立监管路径。有受访者建议创建一个独立的辅料资质认证监管路径,类似于FDA的新型辅料审查试点计划。

来源:药理毒理开发


