您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-11-16 23:00
1.第二类体外诊断设备的产品技术要求中,环境试验还要求吗?
答:按照《医疗器械产品技术要求编写指导原则》相关精神,环境试验可不在产品技术要求中体现。但是相关研究还是应在产品研发过程中体现,并在注册申报时提交相关研究资料。
2.说明书中主要组成成分应包括哪些?
答:应包括最小销售单元中所有参与反应的组分(如各种反应试剂、检测卡)以及检验过程中辅助成分(如干燥剂、比色卡、封板膜、生物安全袋、滴管、校准信息码等)。不含说明书。
3.试剂盒组分单独销售时,应注意什么?
答:销售包装标签应包括完整试剂盒相关信息(如产品名称、注册人、生产地址等),并应注明组分基本信息(包装规格、储存条件及有效期、批号等)以及需注意事项。包装应附原试剂盒说明书。如涉及UDI赋码,应符合相关法规要求。
4.半定量产品的准确度如何要求?
答:可将准确度、检测限合并要求,对于每个分段点均应验证。最低要求为:检测结果与相应参考溶液标示值相差同向不超过一个量级,不得出现反向相差,阳性参考溶液不得出现阴性结果,阴性参考溶液不得出现阳性结果。各量级检测应达到95%的符合率。
5.产品技术要求中,特异性如何要求?
答:产品技术要求中的特异性,一般是指交叉反应,而非干扰。所以一般对于非免疫比浊法的生化试剂无此要求,免疫类产品如有明确的交叉反应物(如不同亚型等)应有此要求。添加的交叉反应物浓度应选择临床上可见的较高水平。
6.瓶间差何时要求?
答:干粉试剂,一般应有瓶间差及复溶稳定性的要求。
7.临床评价时,对于多种样本类型,如何要求?
答:如果申报的试剂产品同时适用于血清、血浆和/或全血样本,那么样本分布应为:血清和血浆可认为属于同一类型进行总量统计(但是需要前期分析性能评估验证两者一致性);如既有血清/血浆,又有全血样本,则应进行同源样本比对(例数按法规要求),且全血正常、异常值的分布要求同血清/血浆。但是血液、尿液、脑脊液等样本之间不适用此情况,应分别进行临床评价。
8.体外诊断试剂包装、标签的设计风格变化、说明书排版和格式的调整,是否需要进行变更注册?
答:不需要。IVD说明书中其他可自行变更的内容参见《总局办公厅关于体外诊断试剂说明书文字性变更有关问题的通知》(食药监办械管〔2016〕117号)。
9.部分肿瘤标志物由三类降为第二类后,说明书中“预期用途”描述如何修改?
答:肿标降类后,申请表中的“预期用途”的写法与常规产品保持一致。说明书中【预期用途】第一段应与申请表中保持一致(同第二类常规产品要求),第二段应以降类文件中调整后的预期用途“临床上用于……”开头,并增加“不用于普通人群的肿瘤筛查”的表述。第二段后续内容与常规产品保持一致,描述相关临床意义即可。
10.申请表中,“预期用途”如何描述?
答:试剂盒应明确“体外”“定性/定量”“样本类型”“被测物”等信息,校准质控品需明确所有项目。举例如下:
例1:用于体外定性检测人血清、血浆中××××的活性。
例2:用于体外定量测定人血清中×××的含量。
例3:与本公司的试剂盒配套使用,用于××检测系统的校准。
例4:与本公司的试剂盒配套使用,用于A、B、C……共×项的室内质量控制。
11.对于超敏C反应蛋白试剂盒,其预期用途中被测物是否为“超敏C反应蛋白”?
答:这种理解是错误的。被测物依然为“C反应蛋白”,“超敏”仅是对于该类产品灵敏度高的一种表述。所以应表述为“用于体外定量检测人血清、血浆中C反应蛋白的含量”。
12.产品按定量还是定性进行申报,需要考虑哪些因素?
答:首先应该考虑临床实际应用需求以及是否有同类产品已上市,其次样本类型也应该考虑,一般对于大便、唾液、咽拭子等非均质化样本很难真正意义上实现“定量”检测。
13.产品想申报家用或患者自测,应该考虑哪些因素?
答:首先应考虑该产品是否有家用或患者自测需求及应用价值,其次应考虑样本获得的方便性,一般应为尿液、唾液、指尖血等。具体要求可参阅《家用体外诊断医疗器械注册技术审查指导原则》。
14.说明书中产品英文名称有何要求?
答:不建议写英文名称,如确需必要,英文名称应当正确、完整,不允许只写缩写。应直译,而非意译。
15.关于产品名称中方法学部分,有何要求?
答:生化试剂盒可参考YY/T 1227-2014《临床化学体外诊断试剂(盒)命名》,不建议使用的方法学表述:终点法、速率法、连续监测法、IFCC法等。胶体金法应为“胶体金免疫层析法”,化学发光法应为“化学发光免疫分析法”或“磁微粒化学发光免疫分析法”。
16.产品包装规格如何规范表述?
答:不同包装规格之间应按照分隔层次分别使用顿号、逗号、分号进行区分,统一以句号结束。如仅单一包装规格,其后可以不加标点符号。举例如下:
例1:48人份/盒
例2:48测试/盒、96 测试 / 盒。
例 3:试剂 1(R1):5mL×5, 试剂 2(R2):5mL×5; 试剂 1(R1):10mL×10,试剂 2(R2):10mL×10。
例 4:反应液:15mL×1, 缓冲液:45mL×1, 校准品:2mL×1。
例 5:××× 型:4mL×10(冻干品,复溶体积)
17.产品技术要求中,线性如何要求?
答:注意区间的表达方式:[A,B]mg/L、(A%,B%);线性下限原则上不应为零,上限应符合临床实际需求。线性偏差可以分段给出绝对偏差和相对偏差要求。设定绝对偏差和相对偏差分界点时,原则上应尽量将医学决定水平浓度置于相对偏差要求区间内;且绝对和相对偏差的要求应考虑分界点的连续性。
举例:医学决定水平20mg/L,偏差要求中分界点为15mg/L,绝对偏差要求≤1.5mg/L,相对偏差要求≤10%。
免疫类产品一般仅要求给出线性区间,可以不要求线性偏差。
18.产品技术要求中,检出限/空白限如何要求?
答:免疫类产品一般有此项要求。可二选一,如果无相应国行标或指导原则,建议给出检出限要求。
19.产品技术要求中,准确度如何要求?
答:如果无相应国行标或指导原则,原则上采用三种方式之一:相对偏差(参考物质或参考方法定值物)、回收试验(人源样本中添加纯品)、比对试验(同类已上市产品)。对于有适用国家标准品的产品,应采用相对偏差方式。
20.产品技术要求中,重复性如何要求?
答:如果无相应国行标或指导原则,可在线性区间范围内,选择2~3个不同浓度水平的样本。浓度选择时,应考虑线性宽度以及医学决定水平,可选正常值和异常值水平(建议为弱阳性)。
21.体外诊断试剂产品申报延续注册时,应如何撰写“产品概述”和“有关产品安全性、有效性主要评价内容”?
答:“产品概述”中建议阐述:产品预期用途;主要组成成分;检测原理;注册证号及有效期。
“有关产品安全性、有效性主要评价内容”中建议阐述:
(1)本次延续注册过程中该产品无变化。
(2)若本次注册证有效期内产品变更情况,应明确变更次数、变更时间及变更事项。
(3)若本次医疗器械注册证有效期内有新的医疗器械强制性标准和/或国家标准品发布实施,为符合新的医疗器械强制性标准和/或国家标准品所做变更情况。
(4)若本次延续注册为注册管理类别有调整的产品,应注明管理类别调整文件,并明确调整前管理类别和调整后管理类别。
(5)若本次延续注册为分类编码有调整的产品,应注明分类编码调整文件,并明确调整前分类编码和调整后分类编码。
(6)若原医疗器械注册证中载明要求继续完成的事项,应说明事项的完成情况。
(7)总结陈述本次延续注册产品安全有效性,符合延续注册要求。
22.目前,化学发光法、胶体金法、荧光法等封闭系统的试剂增加与原注册证说明书中适用仪器不同厂家的适用机型,能否按照《北京市医疗器械快速审评审批办法》第十三条申报快速审评审批?
答:考虑到封闭系统仪器适用性的问题,需要经过充分验证才能证明试剂在新增仪器上的安全有效性,建议注册人增加适用仪器按照正常的“变更注册”流程及材料进行申报。
23.延续注册体外诊断试剂产品为符合新的国家标准品发布实施所做的变化属于无需办理变更注册手续或者无需变化的,应提供哪些申报材料?
答:(1)已注册产品为符合新的国家标准品发布实施所做的变化属于无需办理变更注册手续或者无需变化的说明;
(2)若有无需办理变更注册手续的变化,应提供相关证明材料(至少包括新旧国家标准品说明书复印件和新旧国家标准品说明书涉及条款对比表)。
24.符合《北京市医疗器械快速审评审批办法》第十三条的要求,申报快速审评审批的产品应准备哪些申报材料?
答:
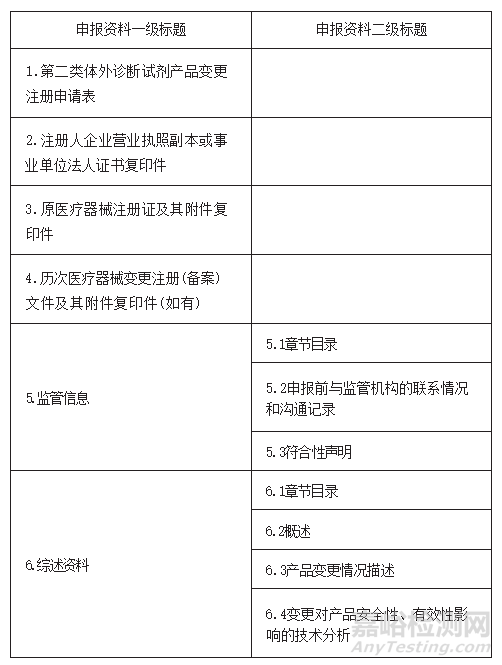
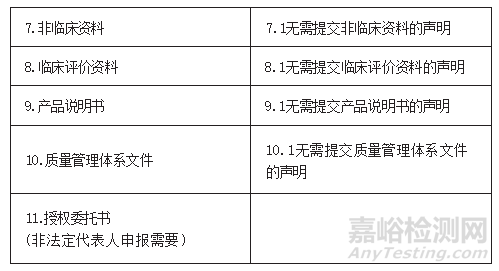
25.按照《北京市医疗器械快速审评审批办法》第十三条申报快速审评审批,增加适用仪器的,对所增加的适用仪器有什么要求?
答:(1)新增适用仪器应与原产品说明书中的适用仪器的自动化程度相同。
(2)新增适用仪器应与原产品说明书中的适用仪器为同类仪器。
(3)新增适用仪器应为国内已上市医疗器械,即已取得医疗器械注册证。
以上三个条件同时满足,才能够按照《北京市医疗器械快速审评审批办法》第十三条申报。
26.由III类降为II类的体外诊断试剂产品,在延续注册过程中可以删除《产品技术要求》中附录里关于主要原材料、生产工艺及半成品要求的相关内容吗?
答:依据《医疗器械产品技术要求编写指导原则》,对于第三类体外诊断试剂类产品,产品技术要求中应以附录形式明确主要原材料、生产工艺要求。对于第二类体外诊断试剂类产品无此要求。

来源:北京市医疗器械审评检查


