您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-10-22 23:07
进口医疗器械代理人管理制度研究
Research on the Management System for China Agent of Imported Medical Device
摘 要
Abstract
目的:对完善进口医疗器械代理人管理制度进行研究。方法:对进口医疗器械代理人的现状采用问卷调查的方式进行摸底,研究进口医疗器械代理人构成、能力、资质等现状,梳理分析进口医疗器械代理人制度运行中的主要问题。结论:目前进口医疗器械通关情形复杂,进口代理人无法掌握全部进口医疗器械信息;进口医疗器械相关主体权利义务不匹配;进口医疗器械代理人监管存在信息不对称。因此,笔者团队建议明确代理人法律责任边界,明确代理人条件和能力,设定规范使代理人可掌握进口产品信息,继续制修订代理人相关管理制度。
Objective: To improvie the management system for China agent of imported medical device. Methods: The current situation of China agent of imported medical device was investigated by means of questionnaires and so on; the composition, ability and qualification of China agent of imported medical device were studied; and the main problems in the implementation of system for China agent of imported medical device were analyzed. Conclusions: At present, the customs clearance of imported medical devices is complicated, and China agent cannot grasp all the information of imported medical devices;the rights and obligations of the relevant subjects of imported medical devices do not match; and there is an information asymmetry in the supervision on China agent of imported medical device. It is recommended to clarify the legal responsibility of China agent as well as their qualifications and capabilities, set regulations to make them to grasp the information of imported products, and continue to develop and revise the relevant system for them.
关键词
Key words
进口医疗器械;代理人;责任
imported medical device; China agent; responsibility
基金项目
2021 年中国药品监督管理研究会课题:进口医疗器械代理人(境外医疗器械注册人、备案人指定的我国境内企业法人)管理制度研究
一、进口医疗器械代理人的管理沿革
第一阶段为2000 年《医疗器械监督管理条例》(以下简称《条例》)发布至2014 年第一次修订期间,此阶段代理人主要承担注册义务。这一阶段中2000年版《条例》中并未就医疗器械代理人①做出相关规定。在2004年版《医疗器械注册管理办法》中规定代理人主要承担注册义务,规定申请境外医疗器械注册的,境外生产企业应当在中国境内指定机构作为其代理人,代理人应当承担相应的法律责任,且在附件中提出了代理人还应当在承诺书中承诺负责报告医疗器械不良事件,并负责与药品监督管理部门联系。
第二阶段为2014 年版《条例》修订开始到2021 年《条例》再次修订,此阶段代理人除注册备案义务外逐渐承担其他义务。这一阶段中,《条例》明确了代理人注册义务,但未明确规定其他管理义务。2014 年版《医疗器械注册管理办法》规定了代理人其他应承担的义务,代理人除办理医疗器械注册或者备案事宜外,还应当承担与相应药品监督管理部门和境外申请人或者备案人的联络;向申请人或者备案人如实、准确传达相关的法规和技术要求;收集上市后医疗器械不良事件信息并反馈境外注册人或者备案人等义务,并在后续的召回和医疗器械不良事件和再评价规范中予以进一步明确。
第三阶段为2021 年《条例》修订后至今,此阶段进一步细化了代理人义务并明确了法律责任。2021 年版《条例》规定向我国境内出口医疗器械的,由其指定的我国境内企业法人向国务院药品监督管理部门提交资料,并要求代理人协助医疗器械注册人、备案人应当履行《条例》第二十条第一款规定的义务。2021 年版《条例》对代理人制度进行了顶层架构,第一次在法规层面明确了代理人注册备案之外的义务,并首次设置了代理人不履行义务的罚则。2021 年版《医疗器械注册与备案管理办法》也明确境外申请人、备案人应当指定中国境内的企业法人作为代理人,办理相关医疗器械注册、备案事项。代理人应当依法协助注册人、备案人履行《条例》规定的相应义务,并协助境外注册人、备案人落实相应法律责任。此外,还进一步明确了国家药品监督管理局应当及时将代理人信息通报代理人所在地省级药品监督管理部门。省级药品监督管理部门对本行政区域内的代理人组织开展日常监督管理。
二、我国进口医疗器械及代理人现状分析
(一)进口医疗器械仍然占据我国高端市场
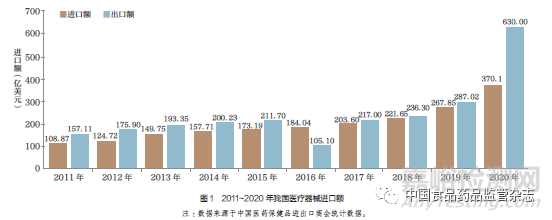
自建立医疗器械注册制度以来,进口医疗器械代理人扮演着重要角色。2011 年至今,我国医疗器械进口额呈逐年上升的趋势,从2011 年的108 亿美元上升到2020 年的370 亿美元, 如图1所示。2021 年1~6 月, 我国医疗器械进口额为171.16 亿美元,同比增长5.96%。
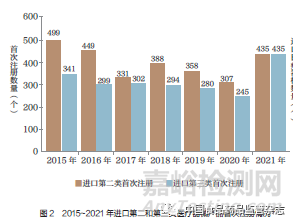
从进口产品注册情况来看,2015~2021 年,进口第二类和第三类医疗器械产品注册量均呈现逐年减少的趋势,2020 年首次注册产品第二类共307 个,第三类为245 个,比2015 年的499 个和341 个减少38.5% 和28.2%,2021 年产品数量有所回升,特别是第三类产品呈现出注册数量新高。进口医疗器械仍然以高端为主[1], 如图2 所示。
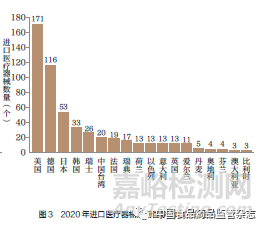
进口来源地仍以美国、德国等国家和地区为主,其中2020年占一半以上,如图3 所示。进口产品中,导管类耗材、体外诊断试剂、高端医疗设备为主要品种。此外,核心零部件、关键原材料进口依赖度仍相对较高。
(二)代理人分布区域集中在上海、北京
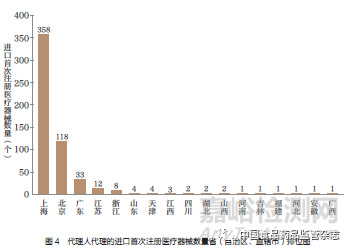
从2020 年进口医疗器械代理人分布情况看,集中在上海、北京、广东等省( 自治区、直辖市),其中上海市代理人代理的进口首次注册医疗器械数量最多,占60% 左右,如图4 所示。目前我国医疗器械外企数量共约2500 家,平均产品注册10.6 件/ 家[3]。从注册证数量排名来看,罗氏诊断、强生医疗器械、西门子、贝克曼库尔特、贝朗、雅培、美敦力、史赛克、波科、库克医疗、奥林巴斯、捷迈、科惠、希森美康、安捷伦、沃芬、徕卡、卡尔蔡司、卡瓦盛邦、施乐辉、奥森多、赛默飞等公司排名靠前。上述企业中,多数在上海注册。
(三)代理人类别多样、管理能力呈现两极分化
笔者团队发放了“进口医疗器械代理人管理制度调查问卷”,收到有效问卷124 份,本文主要根据所有抽样样本调查数据结合企业性质进行分析讨论。根据代理人与注册人、备案人的股权亲疏关系,可分为境外医疗器械注册人、备案人在国内设立的独资子公司,与其有股权关系但并不由其控股的公司,与其无任何股权关系的公司;按照是否经营所代理的产品,又可以分为有医疗器械经营资质和无医疗器械经营资质。代理人与注册人、备案人之间的亲疏关系足以影响代理人能否从注册人、备案人处得到管理支持,代理人是否经营所代理产品也直接影响着代理人是否需要经营企业协助才能履行上市后管理义务。
第一种类型是境外大中型医疗器械公司在境内设立的全资或控股子公司。境外大型医疗器械公司均以在我国境内设立全资的子公司作为代理人为主,这类企业普遍仅代理本企业内的产品,具有医疗器械经营资质,管理能力强,能够覆盖注册、进口、经营、上市后管理全生命周期的义务,能够较好地配合监管部门做好相关工作。经过调研发现,近期因受到“两票制”、集中采购等政策的影响,这类代理人也开始把部分产品的总经销权交由国内大型医疗器械经营公司销售,企业不直接参与销售。
第二种类型是与注册人、备案人无股权关系的境内医疗器械经营企业按经营需求代理产品,并兼顾代理注册和销售。这类公司一般与注册人、备案人没有股权关系,是以经营为目的,其选择合适自己经营的产品分别与不同国家的不同企业谈代理合作,掌握产品实际经销权。这类企业具有医疗器械经营资质,符合《医疗器械经营质量管理规范》相关要求,基本能够满足医疗器械上市后管理的能力。但因代理产品个数和经营能力的限制,相关企业能力上呈现着巨大差异。
第三种类型是与注册人备案人无股权关系,仅从事注册代理,不参与产品经营的企业。通过调研发现这类企业占比不多。这类企业不参与经营,并不符合《医疗器械经营质量管理规范》,在人员构成、设施等方面均无法完全保障。
根据所有抽样样本调查数据显示,自我评价认为有条件和有能力履行法规规定代理人义务的占68.43%,认为尚不完全具备条件和能力的占25.57%,无企业认为自己没有条件和能力。所有企业都认为最难的是实现医疗器械从进口到批发企业的全程追溯,抽样样本中自我评级认为具备追溯能力的占68.86%。第一种类型企业对法规解读跟踪能力较强,85.71% 的第一类受访企业了解代理人义务并知晓不履行义务将依据《条例》九十八条进行处罚。相比之下,第二种和第三种类型受访企业对法规的了解和跟进能力相对较弱, 仅有43.56% 的受访对象表示了解《条例》第九十八条的规定。
由此可见,代理人的管理能力基本按照类别依次递减,代理本集团产品的大型医疗器械企业与仅代理几个产品或仅进行注册代理的企业相比,条件、能力差异较大。
三、代理人管理中存在的问题
(一)进口医疗器械通关情形复杂
根据调研发现,目前进口医疗器械情形多样。从产品角度可大致归为以下几种类型:第一种是已在国内注册或备案,代理自己或授权其他公司经销进口,这类产品由代理人自己或授权经营,进口信息由其掌握。第二种是已在国内注册或备案,但未获授权企业进口,这类产品与“平行进口”的产品相似,可以合法报关进入国内市场。第三种是国外已上市而国内未上市的医疗器械进入国内市场。第四种是国内外均未上市的“三无”医疗器械产品进入国内市场[4]。
在上述类型并存的医疗器械市场中,进口代理人无法掌握全部进口医疗器械信息。一是目前法规虽然规定了代理人代理境外申请人或备案人开展医疗器械注册或者备案工作,但并未限制进口代理人必须是进口总经销商。二是《海关法》相关规定并未限制必须由总经销商或代理人提供医疗器械注册证进行核验[5]。所以进口医疗器械到国内的渠道除了经过总经销商外,还有可能绕过总经销商,以类似“平行进口”[6]的方式由其他渠道辗转进入国内。再者需要尽快检验的产品,在进行合格评定后出具相应的检验证单,进口企业以此为凭证在口岸海关办理进口通关手续[7],并不需要代理人参与其中。三是有部分产品可能会通过走私方式进入国内。以上几种情况都使得代理人无法掌握进口医疗器械流向。
(二)进口医疗器械相关主体权利义务存在不匹配
2021 年版《条例》施行后,代理人应当承担并协助注册人、备案人履行注册、备案,不良事件监测和召回,建立与产品相适应的质量管理体系并保持有效运行,制定上市后研究和风险管控计划并保证有效实施等。这些义务的履行一方面需要得到注册人、备案人的支持,另一方面也需要法规赋予代理人相应的权利以促使代理人有履行义务的能力和条件。
目前,代理人难以完全掌握进口医疗器械产品流向、无法完全履行上市后管理义务, 难以保障进口医疗器械质量管理体系的有效落实。93% 的被调研代理人希望能够增加代理人权限,78.29% 希望需要代理人授权才可进行保税区贴标签等行为,71.43% 希望需要代理人授权才能进口,87.14% 希望要求境外企业告知境内代理人进口销售信息,但有56.71% 的受访代理人认为要求境外企业告知代理人进口销售信息较难实现。
(三)进口医疗器械代理人监管存在信息不对称
如前文所述,代理人并不全部具有医疗器械经营资质,代理人可以经营所代理的产品,也可以不经营所代理的产品,这样的排列组合产生了医疗器械经营监管范畴外的代理人。代理人信息在注册备案时向国家药品监管部门申报,而相关数据库仍需进一步同步到各省级监管部门。2021年版《医疗器械注册与备案管理办法》进一步明确国家药品监督管理局应当及时将代理人信息通报代理人所在地省级药品监督管理部门,省级药品监督管理部门对本行政区域内的代理人组织开展日常监督管理。实务中仍然存在监管部门按照代理人信息前往无法找到代理人或者找到代理人却无法抽样的情况。
四、进口医疗器械代理人管理制度建议
(一)构筑代理人管理制度
1. 厘清代理人与进口注册人、备案人的法律关系
从不同国家和地区的管理模式看,在美国代理人制度中,代理人作为美国监管机构送达信息的对象之一,主要起信息沟通和传达作用。日本的指定上市许可持有人制度并未使用代理字样,但也起到了代理人的作用,其要求指定上市许可持有人承担除沟通之外更多的管理义务和责任,包括产品召回、不良事件监测等。欧盟授权代表制度要求制造商投放到欧盟市场的产品必须同时标有制造商及欧盟授权代表的名称和联络地址,以做好沟通联络,配合建立产品追溯系统,协助做好警戒系统[8],并与进口商和制造商共同承担责任。上述国家和地区之间不同的制度选择均基于各自整套法律制度和现状,如美国的长臂管辖原则使其可以较容易直接追溯到进口的制造商,而欧盟和日本的管理制度近年来则逐步往夯实代理人职责,监管更有可及性的方向调整[9]。
笔者认为,我国进口医疗器械代理人制度应更进一步夯实代理人的职责。进口代理人与境外医疗器械注册人备案人之间首先是一个民事的代理关系,这个代理关系是基于医疗器械监管行政法规的要求而形成的,代理人应当承担代理关系也必须要承担相应义务。代理人本身在医疗器械监管行政法规中因代理人的角色而被赋予的必须要履行的义务,如相关文件中所规定的召回和不良事件监测等。代理人与进口注册人、备案人之间是基于行政监管要求而形成的民事代理关系,代理人又根据行政监管要求而单独需要对所代理的产品承担义务。
2. 构建进口医疗器械代理人管理体系
目前,《条例》中已经就代理人制度进行了顶层设计,随之修订的《医疗器械注册与备案管理办法》和《体外诊断试剂注册与备案管理办法》也在注册备案的阶段进行了规定,前期已经制定的2017 年版《医疗器械召回管理办法》、2019 年版《医疗器械不良事件监测和再评价管理办法》也对代理人需要承担和代为履行的义务进行了规定。这些规章层面的规定分散在各部门规章之中,且相互之间并未达到完全协调和契合。2018 年《进口医疗器械代理人监督管理办法(征求意见稿)》曾公开征求意见,笔者建议应继续加快该办法的制修订工作,将分散在各个规章和规范性文件中对进口代理人的要求进行归集和梳理,明确代理人的责权利,明确进口医疗器械注册人、备案人的责权利。可从以下几个方面进行构建:一是厘清代理人法律责任边界,细化代理人与进口注册人、备案人之间的权利义务和关系;二是明确代理人的条件和能力,确定代理人的基本要求;三是细化代理人掌握进口医疗器械信息的相关制度,从代理人和进口注册人、备案人两个主体角度出发,分别设定管理要求,以便实现代理人履行义务的第一条件“掌握所代理进口产品的信息”;四是明确监督管理要求,在现行法规明确代理人管辖部门的基础上,明确各级监管部门的管理权限和监督模式。鉴于代理人分布集中的特点,笔者认为可在代理人数量较多的上海、北京等地区根据各自的管理经验和要求,探索性地制修订地方管理规范,再进一步在国家管理框架的基础上细化管理措施。
(二)厘清代理人法律责任边界
《条例》明确代理人应当协助注册人、备案人履行第二十条第一款规定的义务;《医疗器械注册与备案管理办法》规定代理人办理相关医疗器械注册、备案事项,并协助境外注册人、备案人落实相应法律责任。但在2017 年版《医疗器械召回管理办法》和2019年版《医疗器械不良事件监测和再评价管理办法》分别要求代理人实施该产品生产企业的调查、召回工作,承担境内销售的进口医疗器械的不良事件监测工作,即不仅是协助还要实际履行。
因此,建议对代理人职责进一步明确和细化,明确释明其法律责任承担的情形。从管理目的考虑,不良事件监测、召回等上市后管理的义务可以由代理人承担和履行,不良事件监测和召回的法律责任也应当由其承担,即使其委托给其他主体履行,也应当由其承担相应责任。质量管理、再评价等也应由代理人进行协助,建议在裁量基准或者规章释义的时候予以释明。除此之外,还应当进一步明确代理人不经营代理产品的情况下,产品相关质量问题的法律后果。
(三)明确代理人条件和能力
代理人应该具备进口医疗器械履行不良事件监测、召回等工作的能力,若其委托他人实施,受托方也应当具备相应能力。代理人还应当具备协助注册人、备案人完成应履行义务的能力。对于产品追溯要求,现行法规和实务中基本以经营作为追溯链,建议应当权衡考虑代理人是否需要掌握经营以外的产品追溯信息。从管理目的来看,代理人只有掌握产品追溯信息,才能进一步履行不良事件监测、召回等义务,故代理人应当具备满足代理产品追溯到进口一级经销商的计算机信息管理系统,以确保产品全流程可追溯。
(四)设定规范使代理人可掌握进口产品信息
现行法规规定进口医疗器械注册人备案人应当委托代理人管理进口医疗器械的追溯、召回等各自上市后管理义务。因此,建议在代理人相关的规定中明确各自权利义务,包括:一是明确进口医疗器械注册人、备案人为其代理人提供进口产品信息的义务。无论代理人作为还是不作为进口经销商,境外注册人、备案人都应当提供境内的卖方、收货、批次等信息。这些信息可以为进口医疗器械代理人协助履行上市后管理义务提供准确的产品流向信息。该措施也可以有效封堵非正常渠道进口医疗器械在国内的流通,并鼓励进口医疗器械注册人、备案人深入理解法规变化,不断配合[10]。
二是建议境外医疗器械注册人、备案人授权委托其他人销售医疗器械的,应当同时获得其进口医疗器械代理人的授权,并与海关部门联合,在进关审查时,除了审查是否具备医疗器械注册证之外,同时审查是否获得进口医疗器械代理人的授权,或经销售进口报关后,需向代理人报备。经销商招投标时,也应提供代理人授权文件。
三是明确进口医疗器械在保税区内加贴中文标签、包装等行为需要注册人、备案人授权代理人进行监督。在保税区内加贴中文标签是进口产品入境的常规做法,严格按照《医疗器械生产质量管理规范》的要求,加贴标签行为属于生产行为的延伸。但基于风险和保税区特殊管理政策,上海等地区的保税区内允许符合《医疗器械生产质量管理规范》(符合ISO13485 :2003《医疗器械 质量管理体系 用于法规的要求》认证)的企业承接贴签等行为,实务中,多数医疗器械产品也采取了贴签模式。调研中发现,大部分代理人能够关注并监督这类行为,但也存在主体通过其他不合规方式进行。建议相关法规应规范进口医疗器械注册人备案人委托国内相关厂商加贴中文标签的行为,要求授权代理人进行监督,对加贴行为进行签字确认。如果未经进口医疗器械代理人确认的,设定相应罚则。这种方法既可以有效规范私自加贴中文标签行为,也可以使得进口医疗器械代理人有效掌握进口产品信息。
四是允许代理人在有条件时查询代理的进口产品信息。给予进口医疗器械代理人查询所代理产品进关情况的路径,代理人可以通过相关系统查询注册证上进口产品报关情况。
(五)明确各级监管部门的监管要求
目前,进口医疗器械产品注册备案的管辖权归属于国家药品监督管理部门,进口代理人的监管管辖权属于地方。《条例》明确进口医疗器械代理人未履行相关义务的,由省级药品监督管理部门责令改正,给予警告,并处罚款;情节严重的还可以处罚到人。《医疗器械注册与备案管理办法》进一步明确国家药品监督管理局应当及时将代理人信息通报代理人所在地省级药品监督管理部门;省级药品监督管理部门对本行政区域内的代理人组织开展日常监督管理。建议应在进口代理人相关管理办法中进一步明确监管要求,可继续采纳《进口医疗器械代理人监督管理办法(征求意见稿)》和现有法规中规定的国家药品监督管理局及时将医疗器械注册证载明的代理人信息通报所在地省级药品监督管理部门;省级药品监督管理部门应当及时收集汇总代理人情况,开展监督检查;省级药品监管部门制定检查频次要求。当按照代理人登记信息无法取得联系的,省级药品监督管理部门报请国家药品监督管理局可以采取暂停进口和销售等措施。如果发生质量问题可以要求代理人进行约谈,并承担与境外企业沟通的义务。
①鉴于2021 年修订的《医疗器械监督管理条例》采用了“境外医疗器械注册人、备案人指定的我国境内企业法人”的表述,但在《医疗器械注册与备案管理办法》《体外诊断试剂注册与备案管理办法》《医疗器械召回管理办法》《医疗器械不良事件监测和再评价管理办法》等规章中仍然使用“代理人”,故本文中仍然使用进口医疗器械代理人(简称代理人)指代“境外医疗器械注册人、备案人指定的我国境内企业法人”。
引用本文
魏俊璟,孙佳斐.进口医疗器械代理人管理制度研究[J].中国食品药品监管.2022.09(224):28-35.

来源:中国食品药品监管杂志


