您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-05-10 11:11
本文中出现的几个术语:
MRA:mutagen risk assessment ,诱变物风险评估。
MIs:mutagenic impurities
PMIs:potentially mutagenic impurities,含有警示结构但未经Ames确证。
降解杂质的诱变风险评估
一、稳定性研究相关的MRA
进行诱变杂质风险评估(MRA),首先是要确定降解途径和降解产物。
强制降解研究
ICH Q1 中描述的设计完善的强制降解实验将会产生一系列的降解产物,这些降解产物的化学结构可以包括在基因杂质的风险评估中。
不过这些降解物属于“潜在的”,因为它们在长期和加速稳定性研究期间可能会形成,也可能不会形成。
加速稳定性研究
加速实验的降解物也应包括在风险评估中。
加速研究可以包括一系列稳定性条件,从露天储存到在最终包装产品中进行的研究。这些条件下形成的降解物归属于“潜在降解产物”的集合中。
长期稳定性研究
长期稳定性储存条件形成的降解物也应包括在MRA中,并归属于“实际降解物”集合中。
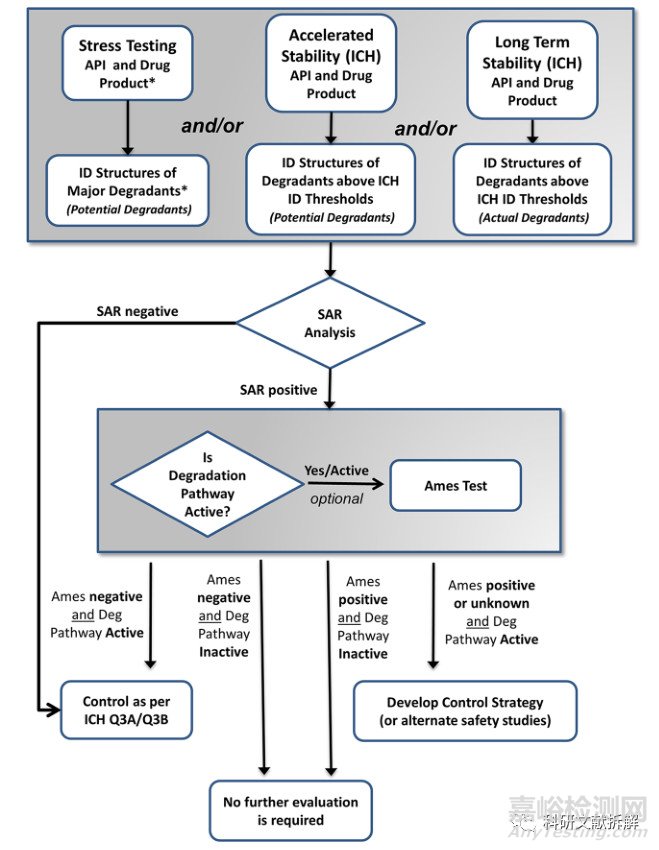
降解产物MRA的一个关键点是和法规要求等价,不能避重就轻,最好也不要过度研究。
应侧重于主要降解途径及其相关的主要降解物;没有在实验中观察到的假设降解物将不需要包括在MRA中。
一旦确定了降解物的结构,就可以通过SAR评估,然后按照M7规则分类。
如果SAR评估为阳性,可以进行Ames实验继续确证。
如果Ames也为阳性,一是通过评估该降解物是否属于药物的活跃降解途径,如果该降解途径非活跃(inactive),该降解物也可以评估后不再进行研究。二是确定在有效期内究竟会产生多少量的MI,通过实际检测值和安全限度值比较。
如果需要,可以制定相应的减缓和控制等相应策略。
使用长期和加速稳定性研究也可以代替Ames实验。
如果该降解物在有效期内的形成水平低于安全限值,则可以不纳入MRA中进行评估。
二、使用软件预测潜在降解
在进行强制降解和相关稳定性研究之前,通过计算机软件预测,有助于考虑潜在的降解途径,并潜在地指导分析方法的开发。
比较常用的商业预测软件是来自Lhasa的Zeneth,Zeneth可以借助数据库中大量的化学原理和专业知识,从不同角度预测药物分子的详细降解产物。
Zeneth的最大优点是可以给出比较全面的降解产物,但同时这也是它的最大缺点。
软件预测一般都会过度预测,很多的产物在实际中不会出现,因此强制降解实验作为补充验证是必不可少的。
三、通过强制降解研究选择鉴定特定降解物
提出了三种强制降解研究的方法:
1:对加速长期产生的降解物进行鉴定
强制降解(在适当水平),以帮助指导加速和长期稳定性研究。
强制降解会导致产生更大数量的降解产物,使降解物更容易通过现有的分析方法进行检测。
强制降解产物结构不一定都要鉴定,除非在加速或长期稳定性研究中也观察到它们。
2:通过特定算法选取降解物进行鉴定
第二种方法利用强制降解研究来描述潜在的降解产物和途径,结构鉴定侧重于“主要”降解产物。
该算法来自于Alsante 等人的研究,将主要降解物定义为:
超过总降解物的10%以上的杂质
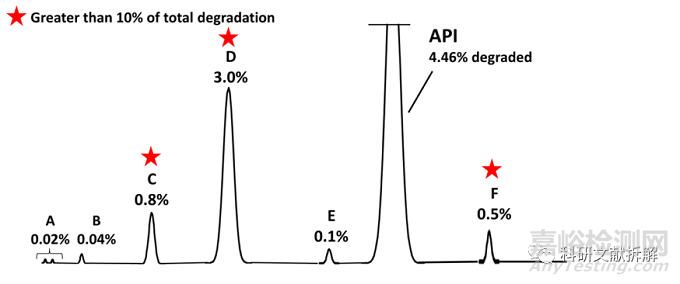
一次降解实验中:API总共降解了4.46%,降解量的10%为0.446%,体系中检测到大于0.446%的杂质有C、D和F,因此该三个杂质为主要降解杂质,需要鉴定结构。
单个最大降解杂质以及大于最大降解杂质25%的降解杂质
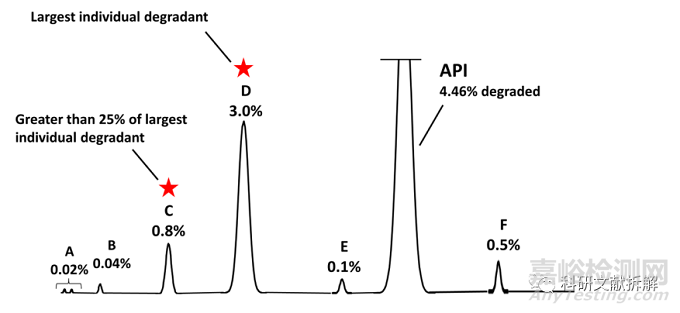
同样一张图,最大单杂为D,D的25%为0.75%。使用该种策略,只需要鉴定杂质C和D。
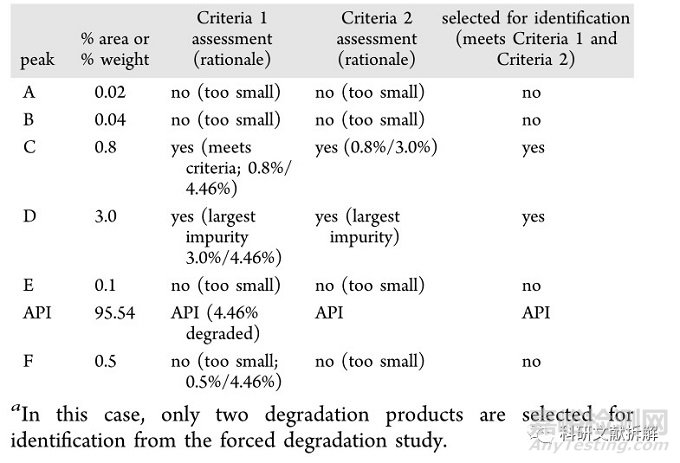
综合两种方案,只需要鉴定两个杂质:
3:通过动力学等效和修正的Q3B阈值来确定降解物阈值
利用动力学等效确定储存周期
通过 Arrhenius equation可以有效预测反应速率和温度之间的关系,因此可以开发更加短期的实验研究数据。
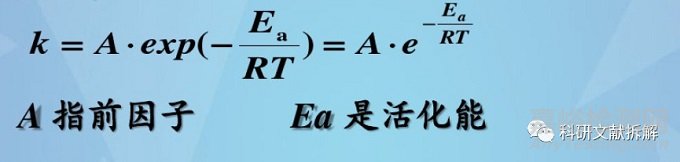
首先要了解药物的降解活化能Ea;如果Ea值未知,根据美国药典推荐的Ea值为19.87kcal/mol(83.14kJ/mol),作为计算平均动态温度的保守方法。
不同Ea值对降解速率影响很大:
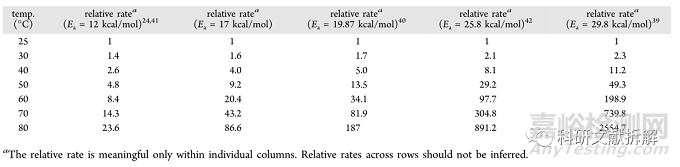
计算在40°C/75%RH下6个月动力学等效所需的天数如下:
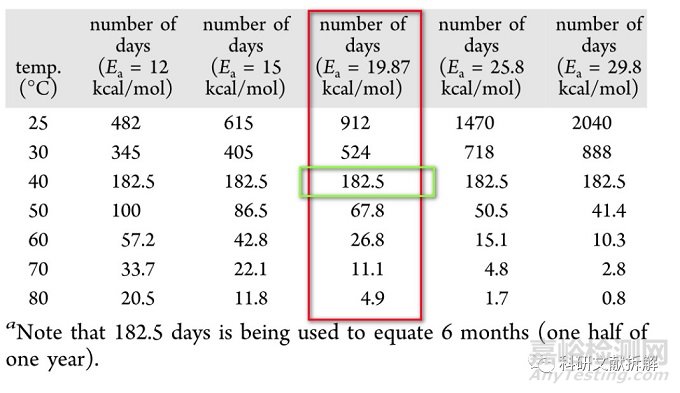
如果温度升高到80℃,可以在4.9天完成40℃时6个月的等效降解。
由于Arrhenius equation没有考虑湿度对降解的影响,因此在不同温度条件下降解研究时,应该保持湿度一致。
强制降解物的鉴定阈值制定
ICH Q1A等法规问答中对诱变降解物鉴定进行了解释:长期稳定性研究中形成的降解物,如果低于ICH鉴定阈值,则不需要对该降解物进行结构鉴定。
根据该条说明,可以制定强制降解实验中的降解物鉴定阈值。
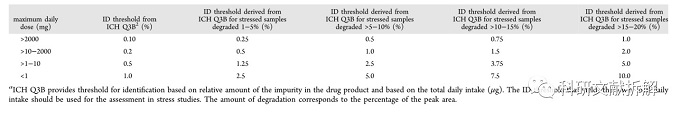
保守估计药物在有效期内降解值为2%,强制降解实验中药物降解率为5~10%,是正常降解的2.5~5倍。因此强制降解实验中的降解物鉴定阈值应该是正常条件的2.5~5倍。
不同降解值对应的ID阈值。
四、案例研究
案例中:
假设降解产物:根据化学机理推测
潜在降解产物:通过强制降解发现的降解产物
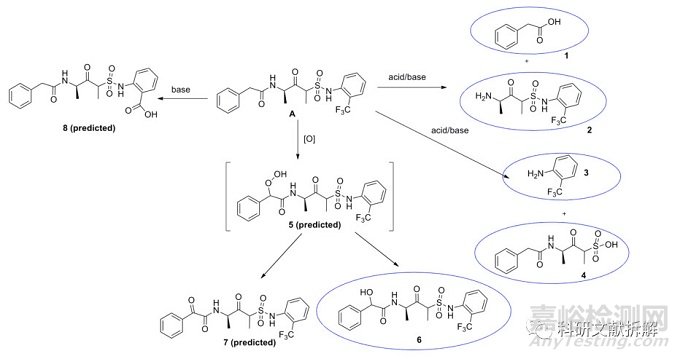
画圈的结构是在稳定性研究中发现超过特定阈值的降解物,并已经被鉴定。
需要进行MRA的降解物
降解物1-4和6应包括在MRA中,因为它们是在强制降解和/或稳定性研究期间观察到的。降解物7和8是假设的降解物,在强制降解期间没有观察到,因此不需要包括在MRA中。
降解物5是6降解途径中的中间体,在强制降解实验中未观察到,根据以上理论5可能不需要包括在MRA中;但是由于API到6的降解途径比较活跃,5是6的中间体,所以需要进一步研究确认。
降解物进行SAR评估
通过评估可以得到降解物3(苯胺结构)和5(过氧化物结构)具有突变性,因此需要对药物在有效期内评估3和5的生成风险。
继续评估有诱变的降解物
降解物3可以在稳定性试验中观察到,但是降解物5没有观察到。
5来源于苄基位置的自氧化,这在药物中是一种普遍的现象,烷基过氧化物通常不稳定,会进一步形成稳定的降解产物,所以在最终产物中无法观察到。
通过RG12915药物的研究数据进行说明:作者在200h发现体系中存在可以测量的氢过氧化物,但是在400h时几乎完全消失,并且在氢过氧化物消失的同时,二次氧化的产物在逐渐增加。因此可以证明中间态的氢过氧化物无法稳定的存在于最终产物中。
化合物5应该包括在风险评估中但是结论是不需要进一步评估研究。
化合物3如果降解水平较高,需要进一步研究,需要开发合适的方法将API水解生成的苯胺3控制到安全水平。
五、其他考虑因素
辅料、包装
基于几十年的长期使用,现有的辅料被认为是人类在医疗产品中使用的安全药物,没有任何重大问题。
虽然辅料本身不存在风险,但是要了解辅料在稳定性以及和药物相容性方面的影响。
可提取物和可浸出物和药品问题相关,可能需要评估。
参考文献:Strategies To Address Mutagenic Impurities Derived from Degradation in Drug Substances and Drug Products
DOI:10.1021/acs.oprd.5b00091

来源:科研文献拆解


