您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2025-03-25 08:42
随着人工智能技术在医疗领域的广泛应用,AI医疗器械/软件的注册成为行业关注的焦点。在医疗器械/软件注册过程中,人工智能(AI)驱动的医疗器械或软件(AIaMD, Artificial Intelligence as a Medical Device)需要遵循特定的注册要求和模板。本文将为您详细解析AI医疗器械/软件注册过程中的关键步骤和注意事项,助力产品顺利上市。
近年来,人工智能(AI)在医疗领域的应用日益广泛,AI医疗器械/软件如雨后春笋般涌现。然而,要想将AI产品推向市场,注册是一道绕不过的门槛。今天,我们就来聊聊AI医疗器械/软件注册的那些事儿。
AI医疗器械/软件注册流程复杂,涉及多个环节:
一、产品基本信息
首先,我们需要明确产品的基本信息,包括产品名称、型号规格、预期用途和分类。这些信息是注册申请的基石,务必准确无误。
二、技术文档
技术文档是注册的核心部分,涵盖产品描述、算法描述、数据管理和软件生命周期等方面。以下为几个关键点:
产品描述:详细阐述AI算法功能、输入输出和运行环境。
算法描述:介绍算法类型、训练数据、开发工具和性能指标。
数据管理:关注数据安全和更新机制。
软件生命周期:遵循相关标准,实施版本控制和风险管理。
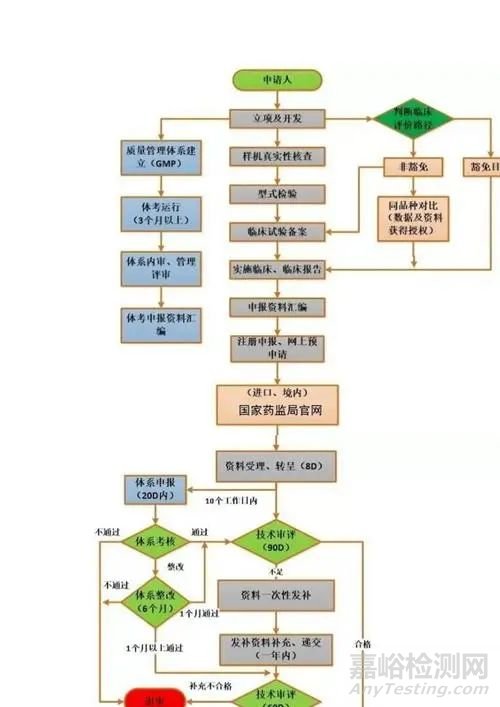
三、临床评价
临床评价报告是证明AI产品安全性和有效性的关键文件。主要包括临床数据、临床意义等内容。临床数据应详述数据集来源、临床验证和性能评估结果。
四、非临床研究
非临床研究主要包括算法验证、软件测试和硬件测试(如适用)。这些研究有助于验证产品的性能和安全性。
五、质量管理体系
建立符合ISO 13485标准的质量管理体系,确保生产过程和不良事件管理的规范性。
六、标签和说明书
标签和说明书是指导用户正确使用产品的重要文件,需明确产品信息、使用方法和警告信息。
七、注册申请表及流程
按照NMPA提供的模板填写注册申请表,并准备相关附加文件。注册流程包括准备资料、提交申请、技术审评、体系核查和审批发证。
八、参考法规和标准
国内法规:《医疗器械监督管理条例》、《医疗器械注册管理办法》、《人工智能医用软件产品分类界定指导原则》等。
国际标准:ISO 13485、IEC 62304、ISO 14971等。
九、注意事项
算法透明度:确保AI算法的可解释性和透明度。
数据合规性:确保训练数据的合法性、合规性和代表性。
持续更新:建立AI软件的持续更新和再评价机制。
注:
ISO 13485是国际标准化组织(ISO)针对医疗器械行业制定的质量管理体系标准。这一标准专门针对医疗器械的特殊性,旨在确保医疗器械在整个生命周期内都能达到最高的质量标准。它不仅涵盖了产品的设计、开发、生产、储存、分发和使用,还涉及了产品的回收、处理和销毁等所有环节。通过遵循ISO 13485标准,医疗器械制造商能够确保其产品的安全性和有效性,从而保障患者的生命安全。
ISO 13485的核心原则是“预防胜于治疗”,这意味着在医疗器械的设计和生产过程中,要考虑到所有可能影响产品质量的因素,并在生产过程中严格控制,以确保医疗器械的高质量。ISO 13485的实施不仅提高了医疗器械的质量水平,也增强了公众对医疗器械安全性的信心。
ISO 13485标准以ISO 9001为基础,但针对医疗器械行业的特殊性进行了调整和增强。它要求在医疗器械产品实现全过程中进行风险管理,并且必须在法规环境下运行。ISO 13485标准的特点包括质量控制、风险管理、合法合规、运营效率、能够追踪和召回产品和设备、工艺和产品改进等。
总的来说,ISO 13485是医疗器械行业中非常重要的质量管理标准,它为医疗器械制造商提供了实施医疗器械指令的框架,并表明了对医疗器械质量和安全准则的承诺。通过实施ISO 13485,企业能够提高和改善管理水平,规避法律风险,增加企业的知名度,提高和保证产品的质量水平,增强产品的竞争力,提高产品的市场占有率。

来源:Internet


