摘 要 / Abstract
目的:为我国药品上市后变更备案标准化管理体系建设提出建议与对策。方法:结合国内外药品上市后变更管理现状,基于科学监管和产业发展的需求,以及药品监管新形势、新要求带来的新机遇和新挑战,参考国外相对成熟的变更管理体系,选择具有代表性的变更情形,结合变更备案工作实践,力求破解药品变更备案实践中监管部门和业界遇到的实际困难和问题。结果:建立分类明确、职责清晰、程序规范的变更管理标准化备案申请填报体系、标准化备案申报资料体系、标准化备案资料审查体系和标准化分级分类监管联动体系。结论:通过建立变更备案标准化体系,指导持有人规范变更、少走弯路,回应公众药品安全关切,助力监管部门和业界更高效、更科学地进行变更管理,服务科学监管,促进医药产业高质量发展。
Objective: This research aims to provide suggestions and strategies for the construction of standardized management system for changes to post-marketing drug records in China. Methods: The study considers the current domestic and overseas status of management system for changes to post-marketing drugs, and the needs of scientific supervision and industry development. New opportunities and challenges brought about by the new situation and requirements of drug supervision are also taken into account with reference to the relatively mature overseas change management systems. Representative examples are selected based on practical difficulties encountered during record changes in order to solve such problems faced by both regulatory authorities and the industry. Results: A standardized record application system for change management is established, featuring clearly defined responsibilities and a standardized procedure. This also includes other systems such as the standardized record submission information system, standardized record information review system, and standardized classification supervision linkage system. Conclusion: By establishing these standardized systems, it is possible not only to prevent marketing authorization holders from taking detours in making changes, but also to effectively address public concerns about drug safety. In addition, this assists the regulatory departments and the industry in carrying out change management efficiently and scientifically to serve scientific supervision, promoting high-quality development of the pharmaceutical industry.
关 键 词 / Key words
药品;上市后;变更备案;标准化研究;实践探索
drugs; post-marketing; record changes; standardized research; practical exploration
近年来,全球医药科技及产业迅猛发展,新技术、新产品、新业态、新模式层出不穷。尽管我国药品从监管科学到科学监管取得了积极进展,但与医药技术和产业高质量发展相比,药品监管科学系统性研究和关键技术的短板仍有凸显[1]。药品上市后变更是监管部门和业界高度关注的热点问题。为加强药品上市后变更管理,2021 年1 月国家药监局出台《药品上市后变更管理办法(试行)》[2](以下简称《变更办法》),国家药监局药审中心相继发布有关技术指导原则,为规范开展药品上市后变更管理提供了工作指南。
但是,《变更办法》及相关技术指导原则未能囊括所有已上市药品变更情形,在具体实施过程中存在某些变更分类模糊,如《已上市中药药学变更研究技术指导原则(试行)》中变更前后质量“不产生明显影响的/ 不产生影响的/基本不产生影响的”在具体品种不同情形的适用性上药品上市许可持有人(以下简称持有人)、资料审查人员及监管检查人员之间可能有不同理解,可能存在持有人变更水平参差不齐、资料审查人员标准尺度把握不一致、监管人员检查重点不统一等问题。这是药品监管部门和业界高度关注和亟需解决的问题。
因此,药品上市后变更备案标准化研究具有迫切的研究需求。笔者参考国外相对成熟的变更管理体系,选择具有代表性的变更情形,力求建立分类明确、职责清晰、程序规范的变更管理标准化备案申请填报体系、标准化备案申报资料体系、标准化备案资料审查体系和标准化分级分类监管联动体系,希望能指导持有人规范变更、少走弯路,同时助力监管部门科学高效地监管,促进医药产业高质量发展。
1、国内外药品上市后变更管理简介
1.1 美国
美国建立了以法规为基础并结合药品科学技术和风险管理原则的变更管理体系,在药品上市前和上市后对变更实行分类明确、职责清晰、程序规范的差异化全生命周期管理模式。
美国食品药品监督管理局(Food and Drug Administration,FDA) 基于上市后变更对药品有效性和安全性相关因素产生不良影响的风险等级, 将上市后变更划分为重大变更、中等变更、微小变更,并递交监管程度不同的报告,即事先批准补充申请(PAS)、即时生效补充申请(CBE-0/CBE-30)和年度报告(AR)3 种形式[3-4]。
同时,FDA 颁布了一系列技术指导原则, 指导企业分别在药品临床试验期、药品上市申请和药品上市后对变更进行分级并开展相关研究。例如, 与药品上市后变更相关的指导原则:《已批准的NDA 或ANDA 变更指南( 修订版1)》[Guidance for Industry Changes to an Approved NDA or ANDA(Revision 1)]、《已批准的NDA或ANDA 变更指南常见问答》(Guidance for Industry Changes to an Approved NDA or ANDA: Questions and Answers)、《可在年度报告中报告的CMC 已批准的生产变更指南草案》(Guidance for Industry CMC Postapproval Manufacturing Changes Reportable in Annual Reports DRAFT GUIDANCE)等。针对不同剂型发布的变更指导原则包括速释口服固体制剂、缓释口服固体制剂及非无菌半固体制剂的扩大规模和上市后变更等[4-9]。
1.2 欧盟
欧盟的药品注册管理制度随着欧盟的发展而发生变化。欧盟是由多个欧洲国家组成的联盟体, 为了减少成员国内部重复的药品审评工作, 统一审评标准,欧盟设立便利、快速的审评体制,灵活的注册模式和廉洁的审评模式。针对药品上市后变更管理, 欧盟主要有如下法律法规:欧洲议会和欧盟理事会指令2009/53/EC《药品上市许可的变更》、欧盟委员会法规EC/1234/2008《关于人用药和兽药上市后变更的审查》、欧盟委员会法规EC/712/2012《关于人用药和兽药上市后变更的审查》( 即EC/1234/2008 法规的修订)。目的是建立一个简单、清晰和更灵活的法律框架,处理医药产品批准后变更,进而更高水平地保护公众健康。欧洲药品管理局(European Medicines Agency,EMA)的上市后变更管理程序与FDA 基本相似,依据变更对产品质量产生影响的可能性将变更分类划分为Ⅰ A、Ⅰ B和Ⅱ类变更[10-11]。
1.3 中国
在我国药品审评审批制度改革之前,我国药品上市后变更的法规体系较不完善,上市后变更监管存在一些弊端。但随着改革的推进,药品上市后变更的监管理念逐步与国际接轨,监管制度不断完善[12]。我国涉及药品变更的法律为《药品管理法》《疫苗管理法》。部门规章为《药品注册管理办法》《药品生产监督管理办法》《变更办法》,并配套出台了《已上市化学药品药学变更研究技术指导原则(试行)》《已上市中药药学变更研究技术指导原则(试行)》《已上市生物制品药学变更研究技术指导原则(试行)》《已上市化学药品和生物制品临床变更技术指导原则》等指导原则来指导持有人开展药品上市后的变更研究[13-16]。
《变更办法》是我国首部专门针对药品上市后变更设置的规范性文件,一方面鼓励持有人运用新生产技术、新方法、新设备、新科技成果,不断改进和优化生产工艺,持续提高药品质量;另一方面,坚决贯彻习近平总书记对于药品监管工作“四个最严”的要求,规范药品变更行为和变更监管,严厉打击非法变更,落实持有人主体责任,保障人民群众用药安全。在药品上市后变更管理方面,我国充分借鉴了欧美上市后变更管理思路,引入了根据风险进行变更管理的理念,同时体现了与国际接轨的监管思路。中国、美国、欧盟对药品上市后变更均是按照变更的程度和风险等级划分管理要求,并分别设立了相应的递交途径、审批时限和变更实施原则。我国将变更级别分为重大变更、中等变更、微小变更。据统计,《变更办法》实施以来有31 个省级药品监管部门发布了《药品上市后变更备案管理实施细则》或沟通交流工作程序等配套文件[17]。
2、药品上市后变更备案面临的机遇与挑战
2.1 新机遇:药品监管新形势、新要求使药品上市后变更常态化
近年来,我国药品监管国际化进程在不断提速,继2017 年6 月原国家食品药品监督管理总局加入国际人用药品注册技术协调会(ICH) 后,2021 年9 月,国家药监局正式申请启动药品检查合作计划(PIC/S)预加入程序。持有人制度的实施进一步优化了资源配置并与国际接轨。结合国际化医药行业快速发展、国际化科学监管需求和持有人制度带来的新机遇,需要运用ICH、FDA、EMA 指导原则和PIC/S相关质量管理规范等科学监管工具[18-20],建立药品上市后变更备案标准化体系,为推动我国药品监管能力提升贡献智慧。
2.2 新挑战:药品变更备案压力更大、要求更高
2.2.1 药品监管部门面临的压力
一是公示时限紧的压力。《变更办法》规定的备案均为告知性备案,备案部门应自备案完成之日起5 日内公示,时限压力大。二是类别判定难的压力。由于药品研发新技术发展和其生命周期管理的复杂性,《变更办法》及其相关指导原则无法囊括所有已上市药品变更情形。对于指导原则尚不明确的变更情形,在如何科学分类、有效防控风险这方面的压力是巨大的。三是审查资料多的压力。在持有人制度下,品种流转激活,变更需求多且不收费,导致企业递交大量变更备案资料。
2.2.2 持有人面临的压力
在制药行业里,随着新设备、新技术、新科技成果陆续落地应用,药物研发能力和已上市药品的质量不断提高。作为变更研究的主体,持有人应及时主动追踪先进的技术要求,主动开展变更研究,在变更中不断提升产品质量[21]。当发生变更时,持有人应当全面掌控变更的起因、过程及结果,实现药品的全生命周期管理[22]。在变更过程中,持有人可能会存在转让品种能否按受让的工艺生产、如何确保药品质量可控、如何开展变更研究等诸多疑惑,质量管理基础薄弱的持有人尤其是由经营企业新转型的持有人,对变更研究不清楚、不知所措、无从下手。
3、药品上市后变更备案标准化研究与实践探索
如何科学地研究分析变更?如何有效地防控变更对药品的安全性、有效性和质量可控性产生的风险?这是药品监管部门和业界高度关注和亟需解决的问题。笔者坚持问题导向,借鉴国内外相对成熟的变更管理体系,选择具有代表性的变更情形,通过信息研究及归纳整理、案例分析、实证研究。力求建立四大标准化体系破解变更备案四大难题,希望能指导持有人规范变更、少走弯路,助力监管部门和业界更科学地进行变更管理,促进药品监管和医药产业高质量发展。
3.1 以填报规范化破解公示信息不完整的问题
《变更办法》实施以来,国家药监局网站新的“境内生产药品备案信息公示”仅显示企业“备案申请内容”,不再显示“备案结论”。由于目前各省级药品监管部门均未发布指导企业如何规范、完整地填报备案申请内容的指南,再加上不同企业变更研究、理解水平参差不齐,部分企业变更填报内容不规范、信息不完整,导致公示内容不被认可,甚至影响招标采购,造成产品上市销售困难。为方便申请人规范合理、简便快捷地填报备案申请内容,让申请人少走弯路,通过建立标准化备案申请填报体系,以填报规范化破解公示信息不完整的问题。
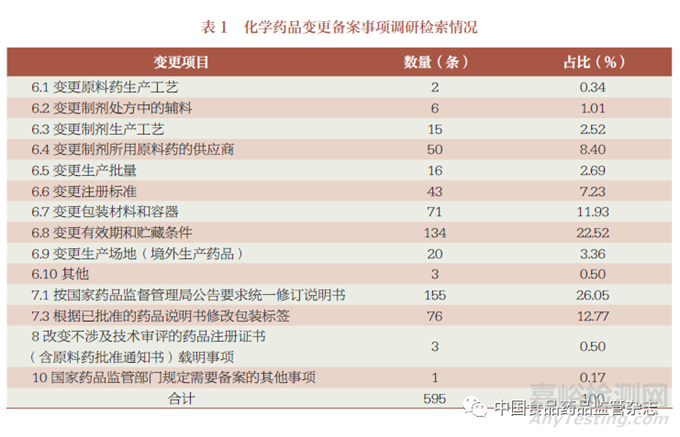
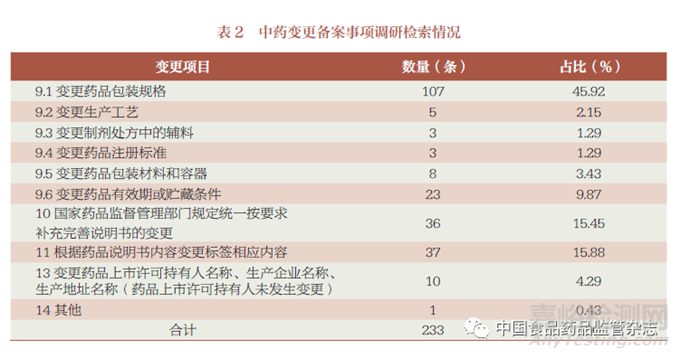
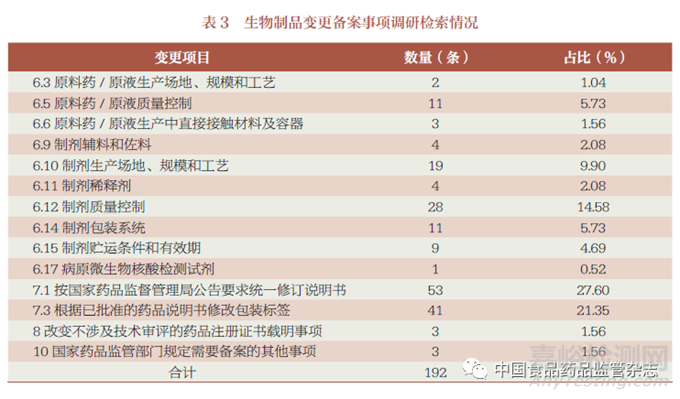
笔者在国家药监局网站“境内生产药品备案信息公示” 公开数据中选择中国化药企业TOP100 排行榜中的前50 家企业、中国中药企业TOP100 排行榜中的前50 家企业、中国生物医药企业TOP20 的企业及2022年生物制品行业上市企业研发投入总额排行榜TOP50 的企业进行备案内容的检索,共整理出化学药品变更备案事项595 条(表1),中药变更备案事项233 条(表2),生物制品变更备案事项192条(表3)。以此为基础,进行分类、提炼,形成化学药品、中药、生物制品的各种变更情形的填报示例。
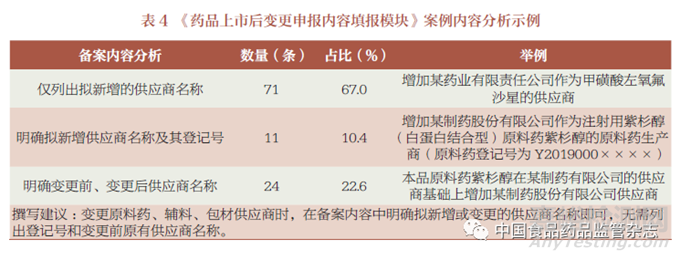
笔者在调研分析、讨论《药品上市后变更申报内容填报模块》时,广泛征求药品监管部门、科研院校、行业协会、持有人、生产企业等多方建议,尽可能阐述清楚变更情形, 同时又能尊重、保护企业商业秘密, 具体示例以化学药品的变更原料药、辅料、包材供应商为例。笔者通过统计, 发现有3 种表述方式, 其中“ 仅列出拟新增的供应商名称”最多,占比67.0%(表4)。经调研分析、讨论后,建议在备案内容中明确拟新增或变更的供应商名称即可, 无需列出登记号和变更前原有供应商名称。不推荐“ 明确拟新增供应商名称及其登记号” 的原因是:同一厂家同一原料药可能有不同工艺的不同登记号,持有人在申报资料中将相关研究情况说明清楚即可,不公示原料药登记号不会影响产品质量,且尊重企业的商业秘密。另外,在企业的档案资料中有原有供应商的情况, 此次变更重点是新增供应商情况, 因此, 可不列原有供应商名称。
3.2 以申报模板化破解企业研究不充分的问题
目前,《变更办法》及相关技术指导原则未能囊括所有已上市药品变更情形,在具体实施过程中存在某些变更分类模糊,某些变更验证评估要求不明确,持有人、资料审查人员及监管检查人员可能有不同理解,存在持有人变更研究水平参差不齐、企业研究不充分的问题。
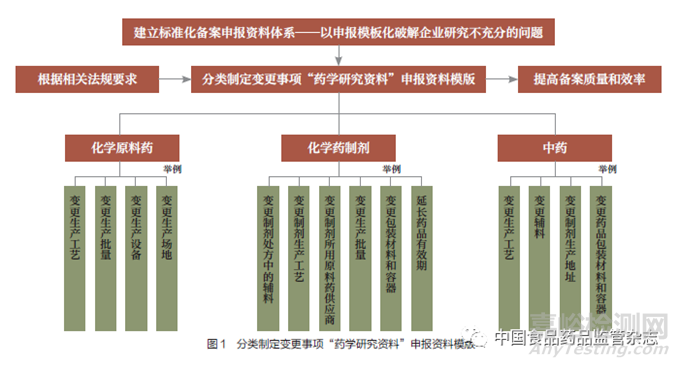
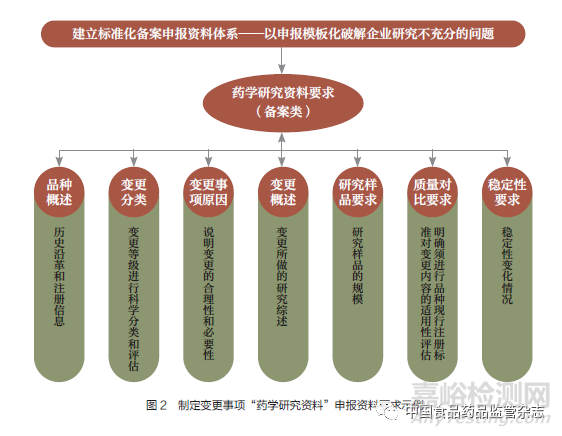
为指导持有人科学、合规开展变更研究,厘清研究思路,根据《变更办法》及相关技术指导原则有关要求,笔者组织开展药品变更模块化文件体系建设研究,中药、化学原料药、化学药制剂备案类变更见图1 和图2。引导企业在备案类变更药学研究工作中有模板可借鉴,指导持有人科学、合规开展变更研究, 厘清研究思路, 提高备案质量和效率。
3.3 以要点标准化破解资料审查尺度不一致的问题
根据《变更办法》要求,自备案完成之日起30 日内完成对备案资料的审查。须建立标准化备案资料审查体系, 用于指导审查人员科学、合规开展药品上市后变更备案资料审查工作,厘清资料审查要点, 规范资料审查行为, 以标准化备案资料审查体系破解资料审查尺度不一致的问题。
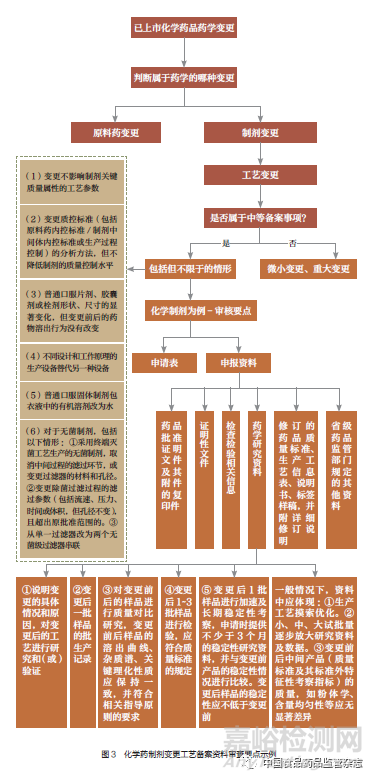
笔者对中药、化学原料药、化学药制剂等备案类变更资料分别制定了相应的资料审查模块,指导审查人员科学、合规开展药品上市后变更备案资料审查工作。通过明确适用范围、审查依据及审查要点,指导审查人员在备案类变更资料审查过程中,遵循科学、合规的要求,避免认识上的混淆、审查上的缺漏,防范变更风险,提高备案质量和效率。建立了各审查模块统一化、标准化的共性审查要点,以化学药制剂变更工艺备案资料审查要点为示例(图3),保证了审查标准的一致性。可指导审查人员紧扣要点,提高审查效率。
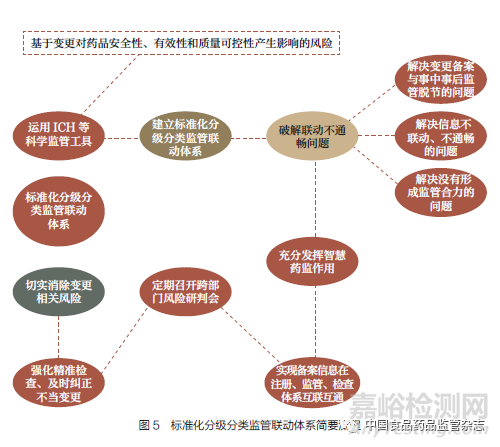
3.4 以信息智能化破解备案监管联动不畅通的问题
《变更办法》及其相关指导原则未囊括所有已上市药品变更情形,而变更情形和问题复杂,事中事后监管脱节、信息不联动、没有形成监管合力问题,需建立标准化分级分类监管联动体系,以标准化分级分类监管联动体系破解备案监管联动不畅通的问题。
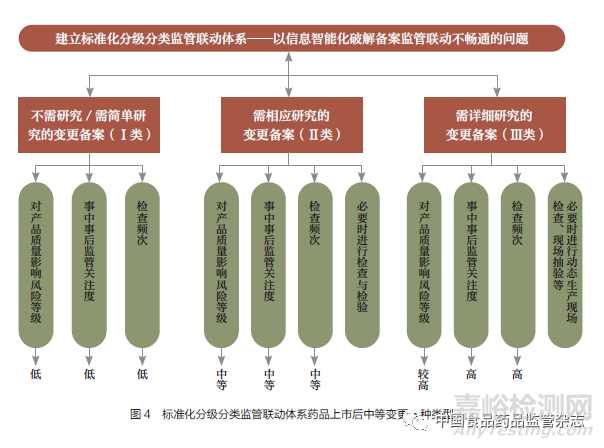
笔者对《变更办法》和相关技术指导原则中已明确、未明确和不能确定的多种中等变更情形,基于上市后变更对药品安全性、有效性和质量可控性产生影响的风险,运用ICH 有关指导原则和PIC/S 相关质量管理规范等科学监管工具,将药品上市后中等变更分为Ⅰ、Ⅱ、Ⅲ 3 种类型,从研究工作要求、风险等级划分、事中事后监管关注度、检查频次、动态生产现场检查、现场抽验等多个维度分别给出对应的监管措施,以期实现提高监管效能、防控药品安全风险(图4)。研究小组按照中药、化学原料药和化学药制剂三大板块,梳理了相应的中等变更分级评估清单,建立了药品变更分类风险评估指标体系和上市后监管联动体系, 分别给出200 余条相应监管建议措施。充分发挥智慧药监作用,将药品备案信息在药品注册、生产监管、检查等系统互联互通,定期组织召开跨部门风险研判会商会,强化精准检查,及时纠正研究、验证不充分或分类不当的变更,切实消除变更相关风险隐患(图5)。
4、结 论
4.1 准确把握变更管理的“变”与“不变”
时刻坚守变更管理的“不变”。一是以人民为中心不变。医药产业发展的终极目标是把公众健康摆在经济利益的前位。二是药品的安全、有效和质量可控不变。从《药品管理法》到《疫苗管理法》,从《药品注册管理办法》到《药品生产监督管理办法》,再到《变更办法》,安全性、有效性和质量可控性的要求高度一致,不断强化变更管理的资料审查、日常检查和有因检查,抓牢质量这个关键。三是法治精神不变。要利剑高悬,加大对变更管理违法违规行为的打击力度,用最严厉的处罚维护法律的尊严。
主动适应变更管理的“变”。一是理念观念要变。强化系统观念和落实“放管服”的理念,不断优化服务,拿出更多变更管理的实招硬招,释放出药品监管和医药产业高质量发展的活力和动力。二是方式方法要变。主动作为,完善有关程序,优化交流方式方法,打通沟通交流“最后一公里”,使企业真正懂变更、会变更、能变更。三是能力水平要变。以新姿态、新能力、新水平来适应新要求、迈步新征程,系统学习、综合提升、全面联动,不断完善药品上市后变更监管体系,最大限度释放监管效能,切实满足人民群众的健康需求 [22]。
4.2 强化变更事中事后监管
强化变更事中事后监管,做好变更现场检查非常重要。资料审查为现场检查提供线索,现场检查为资料审查弥补不足。
严查“为什么要变”。现场检查不仅要关注表面原因,更要关注深层次原因。为什么要变,出发点是什么,是否以牺牲质量的变更来提高利润,特别是关键人员的质量意识和履职情况如何,能否科学判断变更类型,风险防控能力如何。
严查“变了什么”。关注是否有大化小、小化了,是否报小做大,是否随意变更。要特别注意与现场一线操作人员进行深入的交流,通过问细节、问流程,善提问、抓细节,努力了解真实、具体、完整的变更情况。
严查“怎么变的”。要关注变更研究做了哪些,是否充分、科学,记录和数据是否真实、完整、可追溯,记录是谁做的、怎么做的、做得怎么样,特别要注重用系统思维、全链条思维来检查。
严查“变后怎么样”。变更申请后的稳定性考察情况如何,市场抽检和不良反应情况如何,特别关注通过一致性评价和集采品种的变更后质量情况。
4.3 进一步健全完善变更备案标准化体系
笔者团队在中国药品监督管理研究会的支持下,初步建立四大标准化体系破解变更备案四大难题:一是建立标准化备案申请填报体系,以填报规范化破解公示信息不完整的问题。二是建立标准化备案申报资料体系,以申报模板化破解企业研究不充分的问题。三是建立标准化备案资料审查体系,以要点标准化破解资料审查尺度不一致的问题。四是建立标准化分级分类监管联动体系,以信息智能化破解备案监管联动不畅通的问题。通过建立变更备案标准化体系,指导持有人规范变更、少走弯路,同时助力监管部门科学高效地监管,促进医药产业高质量发展。
引用本文
陆才洋,万顺,李超,刘平安*,曾令贵*.药品上市后变更备案标准化研究与实践探索[J].中国食品药品监管,2023(9):22-31.