2016年3月4日,原国家食品药品监管总局发布了化学药品注册分类改革工作方案的公告(2016年第51号)。在公告中,对化学药品的注册分类进行了调整,规定了在境内外均未上市的,在已知活性成分的基础上,对其结构、剂型、处方工艺、给药途径、适应证等进行优化,且具有明显临床优势的药品,属于改良型新药,注册分类为2类[1]。自此,“改良型新药”这个概念正式进入我国药品研发与监管的历史。“改良型新药”类别为业界提供了新的研发路径,其中,2.1类主要针对原料药进行改良(光学异构体、成酯、成盐、改酸根、改碱基、改金属元素、非共价键衍生物),2.2类主要针对制剂进行改良(新剂型、新给药途径、新工艺),2.3类为新复方,2.4类为增加新适应证。与全新靶点和结构的创新药相比,改良型新药具备借鉴已知活性成分药品研究数据的可能性,可缩短临床研发周期,具有成为我国药品研发产业重要组成部分的巨大潜力[2]。
与改良型新药伴随出现的是“具有明显临床优势”这一限定条件。如何理解、研究与判断“具有明显临床优势”是研发改良型新药时需要着力解决的问题。2020年12月31日,药品评审中心发布了《化学药品改良型新药临床试验技术指导原则》,阐述了改良型新药临床优势判断中遵循的原则,以及不同优势的改良型新药的临床试验设计与评价的基本思路。临床优势即患者未被满足的临床需求。在目标适应证中,对比已有的标准治疗,新药或新的治疗手段可显著提高疗效,或在不降低疗效的同时,显著降低当前用药患者的不良反应或用药的相关风险,或显著提高患者用药依从性[3]。
为儿童患者提供安全、有效、质量可控且适用于不同年龄阶段患儿使用的药品是儿童用药开发的目标。在已知活性成分药品基础上优化出具有明显儿童临床优势的改良型新药是拓展儿童应用的常见选择[3]。2021年9月13日,药品评审中心发布了《儿童用化学药品改良型新药临床试验技术指导原则(试行)》,明确鼓励以我国儿科临床需求为导向,根据儿童生长发育特点和儿科临床实践需要,在已知活性成分药品基础上,开发、扩展儿童应用,如增加儿童适应证,或者对已有儿童制剂进行改良,以更好地满足我国儿科临床用药需求,改善儿童给药便利性,提升儿科临床诊疗效果和用药安全。在该指导原则中提出了针对儿童用药的2种常见改良情形,第1种是开发、扩展儿童应用,包括由成人应用扩展至儿童应用及扩展新的儿童应用范围;第2种是改良儿童制剂,包括改变剂型和改变规格。第1种主要对应于2.4类增加新适应证,第2种主要对应于2.2类针对剂型、工艺、给药途径的改良。
本研究重点关注儿童用化学药品改良型新药2.2类临床试验申请的申报与审评情况,基于2016年3月4日至2022年6月30日期间受理的临床试验申请,从申报项目数量、立项通过率、申报治疗领域分布及剂型分布等方面进行了分析。同时,梳理了立项通过和立项未通过的申报项目情况,汇总了立题合理性的关注点,以期为儿童用化学药品改良型新药2.2类临床试验申报提供参考。
一、 儿童用化学药品改良型新药2.2类临床试验申请与审评情况
自 2016 年 3 月 4 日至 2022 年 6 月 30 日,国家药品监督管理局药品审评中心承办的临床试验申请中,有近 300 件按化学药品 2.2 类(或包含 2.2 类)申报,其中 48 件主要针对儿童应用或包含儿童应用(以下称为 48 件 2.2 类儿童用化学药品改良型新药临床试验申请)。
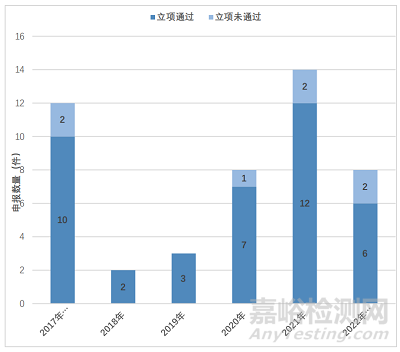
在 48 件 2.2 类儿童用化学药品改良型新药临床试验申请中,2017 年度及之前受理 12 件,2018 年度受理 2 件,2019 年度受理 3 件,2020 年度受理 8 件,2021 年度受理 15 件,2022 年度截至 6 月 30 日前受理 8 件;立项通过(获得《药物临床试验批准通知书》或《药物临床试验批件》)的共 40 件,立项未通过(不予批准或暂停临床)的 7 件(2017 年度及之前 2 件,2020 年度 1 件,2021 年度 2件,2022 年度截至 6 月 30 日前 2 件),企业自行撤回 1 件。(图 1)
图一 2.2类儿童用化学药品改良型新药临床试验申请年度分布情况
Fig.1 Annual Distribution of IND Application for 2.2 Category Modified New Drugs of Peadiatric Drugs
在 48 件 2.2 类儿童用化学药品改良型新药临床试验申请中,有 25 件申请提交了临床试验申请前会议(pre-IND)沟通交流。在立项通过的 40 件申请中,有23 件申请了沟通交流,申请率 57.5%。在立项未通过(暂停临床)的 7 件申请中,有 2 件申请了沟通交流,申请率 28.6%。
在立项通过的 40 件申请中,22 件申报为儿童专用药,18 件申报为儿童可用药(申报适应证同时包括儿童应用和成人应用),其中 8 件列入鼓励研发申报儿童药品清单的品种;境内研发项目申请 25 件,境外研发项目申报国际多中心试验15 件。
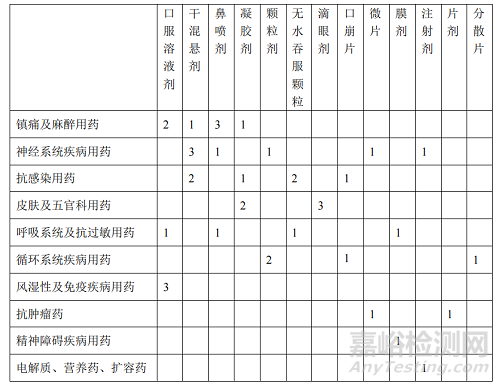
申报共涉及 10 个临床治疗领域,按照临床治疗领域申报项目数量排序,在立项通过的 40 件申请中,镇痛及麻醉用药 7 件(17.5%),神经系统疾病用药 7件(17.5%),抗感染用药 6 件(15%),皮肤及五官科用药 5 件(12.5%),呼吸系统及抗过敏用药 4 件(10%),循环系统疾病用药 4 件(10%),风湿性及免疫疾病用药 3 件(7.5%),抗肿瘤药 2 件(5%),精神障碍疾病用药 1 件(2.5%),电解质、营养药、扩容药 1 件(2.5%)。
申报共涉及 13 个剂型类别,按照剂型类别申报项目数量排序,在立项通过的 40 件申请中,口服溶液剂 6 件(均为儿童专用药),干混悬剂 6 件(2 件儿童专用药,4 件儿童可用药),鼻喷剂 5 件(均为儿童可用药),凝胶剂 4 件(均为儿童专用药),颗粒剂 3 件(2 件儿童专用药,1 件儿童可用药),无水吞服颗粒 3 件(1件儿童专用药,2 件儿童可用药),滴眼剂 3 件(均为儿童专用药),口崩片 2 件(均为儿童可用药),微片 2 件(均为儿童专用药),膜剂 2 件(均为儿童可用药),注射剂 2 件(1 件儿童专用药,1 件儿童可用药),片剂 1 件(为儿童可用药),分散片 1 件(为儿童专用药)。
对立项通过的 40 件申请进行临床治疗领域与剂型类别分布情况的统计,见
下表。
表 1 40 件立项通过申请临床治疗领域与剂型类别分布情况的统计表
Tab.1 Statistical Table of Distribution of Clinical Treatment Fields and Dosage Forms of 40 Approved Applications Items
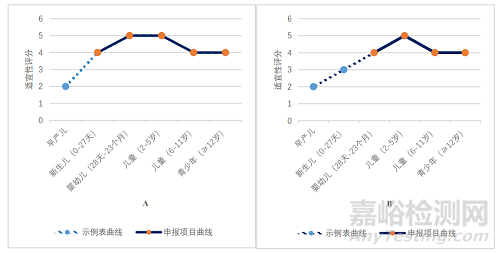
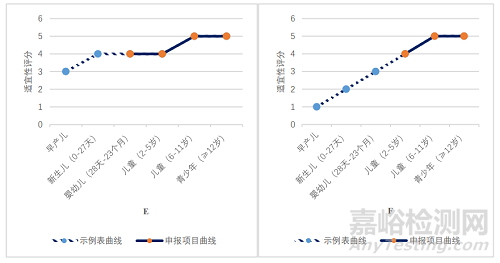
对照 2020 年 12 月 31 日药品评审中心发布的《儿童用药(化学药品)药学开发指导原则(试行)》中附件 1 给药途径/剂型与年龄的关系示例表(以下简称示例表)[10],对立项通过的 40 件申请的剂型/给药途径与年龄(申报适应证中的儿童年龄)的适宜性关系进行统计,如图 2 所示。
图2 40 件立项通过的申请剂型/给药途径与年龄适宜性关系图
Fig.2 Relationship between Dosage Form/route of Administration and Age Suitability of 40 Approved Applications Items
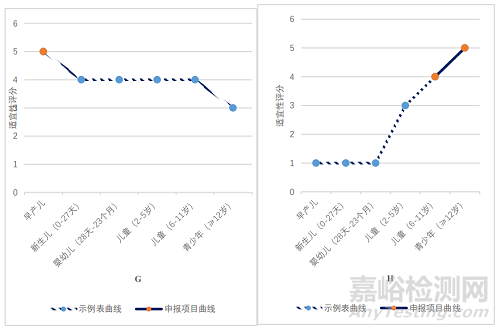
A:口服溶液剂,B:口服混悬剂,C:鼻喷剂,D:凝胶剂,E:滴眼剂,F:口崩片,G:静脉用溶液剂,H:片剂
如图 2A 所示,口服溶液剂在 0 岁新生儿至不满 17 岁青少年年龄段内,适宜性表述为“4,适用性良好”和“5,最适宜和优选剂型”。对照申报情况,在立项通过的 6 件口服溶液剂申请中,申报适应证所涉及的最低起始年龄为 0 岁,最高起始年龄为 6 岁,基本符合给药途径/剂型与年龄的适宜性匹配关系。
如图 2B、C、E 所示,口服混悬剂、鼻喷剂和滴眼剂在出生 28 天以上婴幼儿至不满 17 岁青少年年龄段内,适宜性均表述为“4,适用性良好”和“5,最适宜和优选剂型”。对照申报情况,在获得临床试验许可的 6 件口服混悬剂(包括干混悬剂)申请中,申报适应证所涉及的最低起始年龄为 1 岁,最高起始年龄为 12 岁,基本符合给药途径/剂型与年龄的适宜性匹配关系。在立项通过的 5 件鼻喷剂申请中,申报适应证所涉及的最低起始年龄为出生后 3 月龄,最高起始年龄为 6 岁,基本符合给药途径/剂型与年龄的适宜性匹配关系。在立项通过的 3件滴眼剂申请中,申报适应证所涉及的起始年龄为出生后 3 月龄,基本符合给药途径/剂型与年龄的适宜性匹配关系。
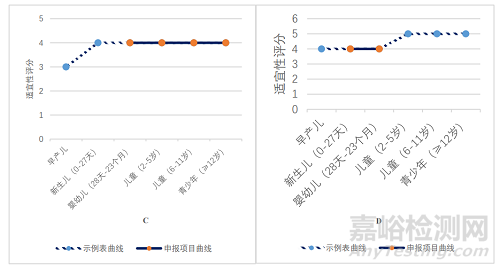
如图 2D 所示,凝胶剂在 0 岁新生儿至不满 2 岁婴幼儿年龄段内,适宜性表述为“4,适用性良好”。对照申报情况,在立项通过的 4 件凝胶剂申请中,申报适应证所涉及的最低起始年龄为 0 岁,最高起始年龄为出生后 4 月龄,基本符合给药途径/剂型与年龄的适宜性匹配关系。
如图 2F 所示,口崩片在 2 岁及以上儿童至不满 17 岁青少年年龄段内,适宜性表述为“4 适用性良好”和“5 最适宜和优选剂型”。对照申报情况,在立项通过的 2 件口崩片申请中,申报适应证所涉及的最低起始年龄为 2 岁,最高起始年龄为 3 岁,基本符合给药途径/剂型与年龄的适宜性匹配关系。
如图 2G 所示,静脉用溶液剂在早产新生儿年龄段,适宜性表述为“5 最适宜和优选剂型”。对照申报情况,在立项通过的 1 件静脉注射剂申请中,申报适应证用于早产新生儿,基本符合给药途径/剂型与年龄的适宜性匹配关系。
如图 2H 所示,片剂在 12 岁及以上青少年年龄段,适宜性表述为“5 最适宜和优选剂型”。对照申报情况,在立项通过的 1 件片剂申请中,申报适应证用于12 岁及以上青少年,基本符合给药途径/剂型与年龄的适宜性匹配关系。
二 、 立题合理性分析及关注点
汇总 48 件 2.2 类儿童用化学药品改良型新药申请,对立题合理性进行分析。在立项通过的 40 件申请中,改良的临床价值判断遵循了《儿童用化学药品改良型新药临床试验技术指导原则(试行)》中提出的以下原则:减少给药创伤或避免侵入性;提高治疗依从性或给药便利性;避免误服、错服、窒息等潜在风险。此3 类是研发者申报与得到审评认可的最常见情形。
在立项未通过的申请中,所涉及的影响立题合理性的考量主要包括以下情形。
(1)剂型与目标治疗儿童年龄段的可接受度不匹配,不符合该年龄段适宜剂型要求。
(2)按照体表面积计算给药剂量的药品,所需剂量范围跨度较大,通过增加更小刻痕单元,提供分割更小给药单元的功能,但仍无法满足儿童患者剂量调整范围的需要,且手动分片后各部分大小不均,无法做到准确定量。
(3)在有已覆盖全年龄段儿童应用、配以刻度量杯提供准确计量条件的某活性成分口服溶液剂上市的情况下,开发其口服滴剂。相比于已上市的口服溶液,在人群年龄段覆盖度方面及计量准确性方面没有额外优势,且由于滴剂给药方式的局限性,给药剂量较大时需要多次取药计量,一定程度上限制了大年龄段儿童和成人应用的便利性。
(4)新剂型改变了释药行为,溶出速度或程度与原剂型不同,导致有效性和/或安全性发生变化。与原剂型相比,新剂型释药速度变慢(非改良目的),药效作用下降,安全性风险增加。
(5)非临床研究结果显示,该活性成分毒性涉及生理发育迟缓和生殖系统的退行性改变,安全范围窄,未见与人体不相关的证据,对儿童受试者存在较大风险。
(6)已有同活性成分药品开展过儿童临床研究,且研究数据显示该活性成分对儿童生长发育有潜在影响。
(7)根据我国对同活性成分或含有同活性成分药品潜在风险特征的监测数据,该类药品已被实施管控或撤市处理,不同意开发同活性成分或含有同活性成分的新剂型。
三 、 讨论
改良型新药为满足我国儿科临床用药需求提供了可行路径。我国儿科临床中存在着“无药可用”和“有药难用”的问题。部分境内外均已上市的药品,仅在境外获得了儿童应用许可,而我国长期没有企业进行儿童应用许可的申报,无法满足我国患儿用药需求。部分批准了儿童应用的药品,缺少儿童适宜的剂型或规格,造成剂量不准确,使用便利性差等问题。在已批准的成人适应证基础上进行改良,开发儿童应用,或者将适应证由大龄儿童向低龄儿童扩展,以及为满足儿童应用目的,改变剂型、规格等制剂特征,均属于 2 类改良型新药研发范畴。从目前统计数据看,研发者对于 2.2 类制剂改良的关注度较高,立项申报及审评通过率稳定增长,而对于申报扩展适应证范围的 2.4 类热情有限。分析原因,可能与研发者对儿科未满足的临床用药需求的了解有限或与预期 2.4 类研发投入更有关。未来,药品监管机构应着力对 2.4 类改良型新药研发加强引导,对其所涉及的临床研究策略给予清晰说明。
在考虑针对儿童使用开发新剂型产品时,重点需要关注新剂型在目标治疗儿童中使用的适宜性,充分保证或提高儿童患者的依从性,以及满足其他特殊要求,以获得积极的获益风险比[4]。从目前统计数据看,立项通过的 2.2 类改良型新药与《儿童用药(化学药品)药学开发指导原则(试行)》附件 1 给药途径/剂型与年龄关系示例表的匹配度较好。口服溶液剂、口服混悬剂、鼻喷剂、凝胶剂、滴眼剂、口崩片、注射剂(静脉)和片剂所申报的目标治疗儿童年龄段均与示例表适宜性表述“4,适用性良好”和“5,最适宜和优选剂型”匹配。良好匹配度一方面体现出了药品技术审评参照指导原则进行标准把控的一致性,另一方面,也说明研发者对于指导原则要求的理解与认可。需要注意的是,指导原则示例表仅作为研发者立项及审评决策的参考,在立项评估中还需充分考虑我国儿科临床需求的动态变化。另外,一些较为新颖的剂型并未包含在指导原则的适宜性示例中,无水吞服颗粒、微片、膜剂等,增加了研发者立项难度,也对审评人员的评价水平提出了更高的要求。
制剂改良特征与临床优势定位的吻合是立项通过的关键。在立项前,需深入了解儿童患者人群的病理生理特点、诊疗条件和用药习惯,同时需充分调研比较临床上已有治疗手段或药品的使用现状,以及临床疗效、安全性、便利性等特征,最终,综合分析改良后的产品是否能够为儿童患者带来更多的获益,填补未被满足的临床需求。另外,对于活性成分药品相关监管环境的变化、临床诊疗指南信息的调整也应有所了解,及时调整研发策略。有时,对预期临床优势的描述(如提高依从性)并不能替代临床研究证据,仍需在必要的临床试验中设计合理指标对预期临床优势进行证明。研发者需要树立“临床优势的评价应贯穿改良型新药研究的全过程”的意识,在临床研究阶段,研究目的与试验设计应始终围绕评价改良特征及其所预期的临床优势,用试验数据体现改良特征并证明临床优势。值得注意的是,申报前的沟通交流在提高立项通过率方面发挥了重要作用。从目前统计数据看,立项通过率与沟通交流申请率具有相关性。在沟通交流过程中,研发者可以得到有关立题合理性及临床研究计划等方面的建议意见,对于明确临床需求信息,确立科学合理的改良目标,提高研发效率,减少无效申报,具有积极意义。
四 、 结语
纵观药品研发历史,制剂改良相关的技术革新曾创造出一个又一个高光时刻。如果说,化合物创新与改造是以疾病病因与发病机制源头为切入点,那么,针对制剂的改良则更侧重人们使用药品时的实际需要与优质体验,创伤更小、感觉更舒适、操作更简单等等。采用创新的技术方法,帮助拓展临床用药途径,优化特殊人群给药方案,是有重要价值的研发方向。
受篇幅所限,本研究仅对儿童用化学药品改良型新药临床试验申报与审评情况进行了描述性分析,尚未做具体问题的深入挖掘与整理,比如按照《儿童用化学药品改良型新药临床试验技术指导原则(试行)》中“多目标同时优化制剂特征”的改良建议,统计分析在增加儿童适应证的同时开发适宜该年龄段患儿使用的适宜剂型和适宜规格的情况,或者,针对多家企业申报同一适应证的同剂型品种的情况,以及在呼吸、镇静相关适应证中申报率较高,而在其他一些治疗领域却没有改良制剂申报的情况进行分析。未来,随着儿童用化学药品改良型新药申报案例的积累与审评经验的丰富,会持续关注此类申报情况,定期进行更细致的梳理和更深入的分析,供研发者与监管者参考。