您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-08-04 13:20
药品杂质作为影响药品安全有效性的关键因素,在药品研究、质控过程中发挥了重要作用。本人从事多年的药物杂质分离纯化工作,本文将重点讨论药物杂质对照品的制备获取方法,包括以下三部分内容:药物杂质对照品粗品来源;药物杂质对照品纯化分离方法;药物杂质对照品制备获得方法。文末将以实例分享制备获取过程。
1 药物杂质对照品粗品来源
药物杂质可以分为工艺杂质、降解杂质、从起始原料或试剂中带入的杂质等。由于药物杂质含量往往较低,无论从中间体、反应液或母液提取难度大,获取成本高,因此快速获取高含量的杂质粗品尤为关键。对于中间体或成品杂质,一般参考合成工艺路线,进行破坏性试验,例如改变反应物料的配比、反应温度、后处理方式等。或者参考药物稳定性影响因素试验,采用高温(例如60℃~100℃)、高湿(例如RH90%±5%)、酸碱(例如0.1mol/L-1.0mol/L的盐酸或氢氧化钠)、氧化(例如0.1%到3%的过氧化氢)光照等剧烈条件来提高对反应物的破坏降解,提高杂质含量。另外也可以采用定向合成方法,即需要先反推杂质结构,再而设计相关合成路线,待分离纯化后,再经液质分析确认。
小结:药物杂质对照品粗品来源是制备分离的关键,杂质含量低,分离难度大,时间成本高。优选定向合成方法,次选破坏性试验法,以提高杂质含量为目的。特殊情况下,对于某些杂质无法推导其可能结构或者经破坏性试验杂质含量仍较低者,只能通过富集法。
2药物杂质对照品纯化分离方法
药物杂质对照品纯化分离方法通常包括:结晶法、柱层析法、制备薄层色谱法、制备色谱法和成盐法、酸碱法等。由于药物杂质对照品粗品的获得过程,往往含量较低。因而本文主要讨论制备色谱法在分离纯化中的应用。
2.1 结晶法
常用的结晶法有冷却结晶法和反溶剂结晶法。冷却结晶法是根据物质在某一溶剂不同温度下溶解度不同,将待纯化物加热至溶解后,降温析晶的过程。冷却结晶关键在于溶剂的选择,通常根据“相似相容”原理。而反溶剂结晶法指的是选取两种溶剂(良性溶剂和不良溶剂),将待纯化产物溶于良性溶剂再加入不良溶剂析晶的过程。结晶法主要适用于杂质含量比较高的产品(例如定向合成),本文不做重点讨论。
2.2 柱层析法
柱层析法即常说的“过柱子”,分离原理是根据物质在固定相上的吸附能力不同而进行分离,常用的吸附固定相是硅胶和氧化铝,分离的关键在于装柱方法(湿法和干法),上样方法(湿法和干法)和洗脱剂的选择(溶剂选择和洗脱方式)。柱层析法具有能耗低、洗脱剂易于回收等优点,同时也存在分离过程费时、效率低,对于复杂化合物纯化效果差等缺点。
2.3 制备薄层色谱法
制备薄层色谱法(爬大板法)可以快速分离反应体系混合物的多种成分,是一种实用快速获取少量对照品的方法。制备薄层色谱法操作流程主要包括:硅胶板的选择(通常自制、厚度好)、样品上样、洗脱展开、色带检测、刮板溶解。制备薄层色谱法虽然操作方便,设备简单,但仍存在样品处理量小的缺点。
2.4 制备液相色谱法
制备液相色谱法是一种具有高灵敏度、高选择性的快速分离技术,其目的是对产品进行快速分离纯化。在纯度、收率、分离效率等方面远优于传统的制备方法,已广泛地应用于药物研究的分离和纯化。
2.4.1制备液相色谱和分析液相色谱的比较
制备液相色谱法分为正相制备色谱和反相制备色谱两种。其中反相制备色谱使用广泛,固定相一般采用反相填料(C18、C8)等,药物的分离纯化多采用以C18为填料的反相色谱柱。与分析液相用于对样品进行定量和定性不同,制备液相的目的是对特定组分进行分离和纯化,不需要很高的精密度和准确度。
2.4.2制备液相色谱的进样操作
制备液相为了提高制备量,经常采用超载进样。超载方式有两种,一种是“浓度超载”,即待纯化物质溶解于较小的进样体积,提高进样浓度;另一种是“体积超载”,即将待纯化物质溶解于较大量的进样体积,减少进样浓度。浓度超载和体积超载都将导致化合物分离度的降低。样品在流动相中具有足够溶解度情况下,优选浓度超载法。通过建立合适的制备方法分离化合物峰,经过检测器时被检测到并收集起来,进而通过蒸发或冷冻干燥除去流动相而得到纯品。
2.4.3制备液相色谱方法的建立和注意事项
2.4.3.1了解待纯化样品的相关物性
了解杂质粗品的来源,通过定向合成、破坏降解或母液浓缩富集,初步了解其中可能含有的化合物。利用液质联用技术,检测分析样品的分子量和分离情况。对于经酸碱破坏降解的杂质粗品液,可以通过比较不同时间段,放置于不同环境温度,考察目标杂质含量变化情况,了解杂质的酸碱稳定性,热稳定性,为后期去除流动相提供基础理论。
2.4.3.2 待纯化样品的预处理
杂质样品往往来源复杂,含有不可逆吸附作用物质,一方面容易堵塞污染柱子,另一方面降低柱使用寿命,影响重现性。因而,必要的前处理如萃取、过滤、析晶、等分离方法尤为重要。另外,关注分离体系是否含有重金属类物质,样品溶液进样是否需要调节Ph等。
2.4.3.3 建立初步制备分离方法
根据目标化合物结构,推测其极性大小,采用等度或梯度洗脱方法摸索保留时间。关注待分离目标化合物与其前后杂质的分离度,而不要求在分析柱上得到所有组分的基线分离。流动相中水相调节剂常加入:甲酸、乙酸、甲酸铵、乙酸铵等,有机相常使用乙腈和甲醇。制备分离方法的初步建立,在于摸索从分析方法到制备方法的区别,需要确保所有杂质全部洗脱。
2.4.3.4 制备方法的优化
优化方向的目的:回收率和纯度。建立了初始的制备方法后,确定了目标杂质峰的位置以及整个洗脱体系的情况后,为了节约时间和成本,我们只关注目标杂质和相邻杂质的分离情况,而经常采取浓度超载或体积超载进样。制备中常常因为超载而无法使目的物与邻近杂质间达到完全基线分离,可以优化出峰时间,采用分段收集的方法得到指定纯度的目的物。对于体系复杂者,可以二次制备纯化。
3药物杂质对照品制备获得方法
对于定向合成方法,往往能得到较高含量的目标杂质,这种情况下,通常采用萃取法或结晶法,减压浓缩除去溶剂或过滤洗涤烘干,即可得目标杂质对照品。需要注意的是,杂质对照品往往不稳定,尽量快速低温制备烘干,最后保存于低温冰箱。通过柱层析法或制备薄层色谱法分离的杂质,通常也是低温快速减压浓缩至干,低温冰箱保存。而对于含量较低的杂质粗品而言,通过制备液相色谱法得到的制备液。对于弱极性物质,可以先减压去除乙腈,再萃取浓缩至干;对于水溶性强的物质,可以采用不含盐的水相-有机相再次制备除去缓冲盐,最后采取低温旋除水或冷冻干燥法去除水,即可得到目标杂质。
实例分享:含有β-内酰胺类抗生素开环物的制备获取过程
开环物杂质溶液获取方法
根据文献,可知β-内酰胺是指一类含有四元内酰胺环的有机结构,在酸碱条件下容易开环。因而优选酸-碱破坏法(0.1mol/L-1.0mol/L盐酸或氢氧化钠水溶液),调整物料比和反应时间(减少副产物例如聚合物的产生)即可得含量较高的杂质溶液;
开环物杂质溶液的纯化分离方法
得到开环物杂质溶液后,需要调节反应液Ph至中性左右,以终止反应。因而体系中引入了无机盐和其他可能杂质。开环物含有羧基和仲氨基,易溶于水。纯化分离优选反相制备色谱,一方面可以出去可溶性盐,另一方面可以进一步提高纯度。
反相制备色谱方法的建立
以分析液相(4.6*250mm-5μm)为例,可检测目标杂质的极性和相邻杂质情况,制备方法的建立以21.2*250mm-5μm制备柱为例,流动相以纯化水和乙腈为例,以平行放大原理,
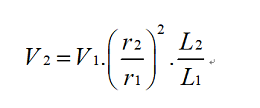
其中,V2、r2、L2分别为制备色谱的流量、内径、柱长;V1、r1、L1分别为分析色谱的流量、内径、柱长;建立初始方法,然后超载进样,优化方法,收集制备液,经液相分析,纯度满足后,连续进样,得到不含盐的水相制备液。
水相制备液的后处理
得到符合纯度要求的水相制备液后,通常先低温旋除有机溶剂,然后依据杂质稳定性,选择低温浓缩至干或冷冻干燥,即得杂质对照品。最后低温冰箱密封保存。
总结:药物杂质对照品在质量研究中不可或缺。然而由于含量低,获取难等原因,目前市场上杂质对照品价格昂贵,增大药企研发成本。制备液相色谱在纯化分离有着显著的优势。

来源:药事纵横


