您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-06-29 19:40
2017年6月加入国际人用药品注册技术协调会(ICH)后,中国药品研发与注册申报开始与国际接轨。尤其在面临国内集采政策和医保谈判降价的巨大压力下,仅局限于国内市场,将面临研发投入无法收回的困境。走国际化路线,参与全球竞争,是我国制药企业发展的一个必然趋势。
中国企业的“出海”热潮如火如荼,而美国是全球药品监管水平最高且销售额最高的国家,中美双报已经成为了大型药企注册申报的标配模式,中国制药企业在FDA的药品注册数量呈逐年递增趋势。本文将针对美国药品的注册分类、审评时限及费用、审评流程、缺陷信及年度维护,分别进行专项解读。
一、美国药品注册分类
美国药品注册主要包括以下分类:
IND (Investigational New Drug)--新药临床试验研究申请
NDA (New Drug Application)--新药上市申请
ANDA (Abbreviated New Drug Application)--仿制药申请
BLA (Biologic License Application)--生物新药或生物类似药申请
OTC (Over-the-Counter Drugs)--非处方药
DMF (Drug Master File)—药品主文件(包括4类)
II类:原料药、原料药中间产品等;
III类:包装材料;
IV类:辅料,着色剂,香精等;
V类:其他,包括无菌设施、生产工艺等,经FDA同意方可申报。
二、审评时限及费用
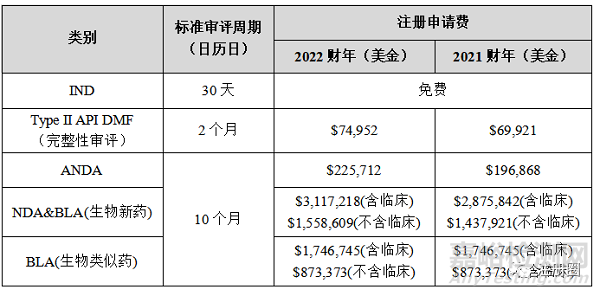
不同类型的药品注册,FDA的标准审评周期自30天至10个月不等。此外FDA有四条加快创新药品上市的特殊审批通道:突破性疗法(Breakthrough)、优先审评(Priority review)、快速通道(Fast track)、加速批准(Accelerated approval)。相较于国内,FDA的审评速度较快,拿到FDA的立卷接收函或批准函,对于加速国内药品审评和增进信任意义重大。
FDA官网会定期公布使用者付费信息,较受关注的有:《处方药使用者付费法案》(PDUFA)、《仿制药使用者付费法案》(GDUFA),《生物类似药使用者付费法案》(BsUFA)。各类药品的注册申请费基本呈逐年递增趋势,高效注册申报可为药企节约大量时间和金钱成本。
三、审评流程
IND
审评周期为30个自然日,随时有收到缺陷信的可能,并要求在短期内完成回复,完成回复后会收到FDA批准进入临床的信函。30天内不能完成缺陷回复,会进入临床暂缓阶段。
DMF
行政信息审评:申报资料递交后,FDA会进行行政信息审评,合格后将被列入DMF清单(List of Drug Master Files),状态为A。行政信息若有缺陷,FDA会通知申报方进行补充,缺陷解决前该DMF无法被引用。
完整性审评:对于II类原料药,完成交费后,FDA开始完整性审评。审评合格将被列入另一个“可引用清单(Available for Reference List)”。审评不合格,申报方会收到FDA发布的缺陷信,并要求在限定期限内完成回复。
技术审评:只有被制剂产品引用后,FDA会启动技术审评,用于支持其特定用途(如用于生产固体制剂或注射剂的原料药)。该DMF持有者随时会因为制剂产品的初次上报、变更等收到相应缺陷信。DMF不存在批准的说法。
ANDA、NDA、BLA
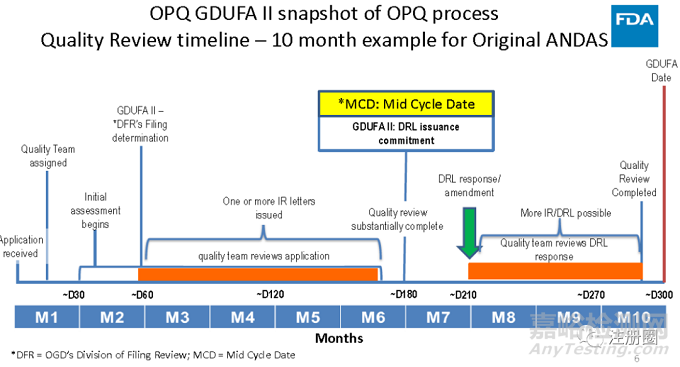
当轮审评周期内,FDA会进行三个阶段的申报资料审评,分别是形式审评,技术审评及缺陷信回复稿审评。结合批准前现场检查的结果,FDA会做出是否批准产品的决定。详细信息见下:
阶段1:
形式审评。一般在60个自然日内,FDA会完成形式审核确定是否立卷。若有少量微小缺陷,FDA会发布IR (信息请求函),并要求7日内回复(若逾期回复IR或回复稿不被认可,FDA会拒收产品);若有重大缺陷或大量微小缺陷,FDA会严格按照拒收标准拒收产品注册文件。
被拒收的产品,申报费用不会全额退返,对于时间和金钱成本都是巨大的浪费。深度解读美国法规,合理制定申报策略,可以为药品开发降本提速。
阶段2:
技术审评。在审评周期的中期左右,FDA不同领域的专家(临床、非临床、CMC等),会相继完成申报资料的初步审核,并发布IR (信息请求函)或DRL(学科审评函),要求申报方在限定日期内,完成缺陷回复。
阶段3:
IR,DRL回复稿审评。FDA会尽量当轮完成IR,DRL回复稿的审核,结合批准前现场检查的结果,做出是否批准产品的决定。
若申报方逾期回复IR,DRL缺陷信,或回复稿不被FDA认可,在审评周期结束时,FDA会拒绝批准该产品,并发布CRL(完全回应函)。CRL中汇总了各学科待整改的所有缺陷,并根据缺陷严重程度,要求申报方在限定日期内完成缺陷回复,产品申报资料将进入下一轮审评周期。
若产品各学科的申报资料均完整合规,且通过现场检查,FDA会发布批准信或临时批准信(专利因素等)。
下图是美国仿制药审评时间轴,清晰呈现了上述三个阶段的审评流程,具有良好的指导意义。
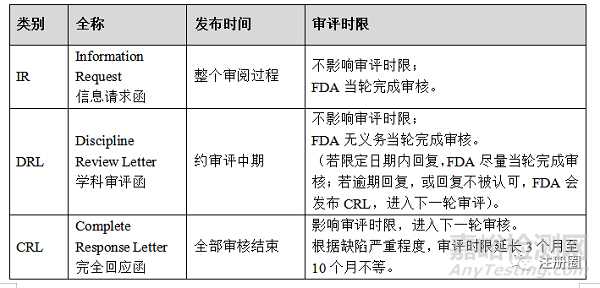
四、缺陷信分类
据上文可知,在审评的不同阶段,FDA会发布三种类型的缺陷信。IR及DRL缺陷信不会影响产品审评时限,而CRL缺陷信会延长产品审评时限。
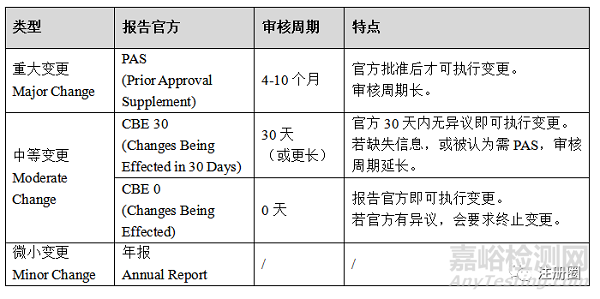
五、年度维护
IND: 临床方案修订、信息修订、临床安全性报告、年报、IR回复等。
DMF: 年报和变更修订等。
ANDA、NDA、BLA:年报和变更修订等。
药品上市后变更管理是全生命周期的重要部分。药品发生变更后,需通过全面的研究工作,考察评估该变更对药品安全性、有效性和质量可控性的影响程度,包括化学、物理学、微生物学、生物学、生物等效性、稳定性等。
来源:注册圈


