您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2022-03-20 23:21
摘 要Abstract
新修订《药品管理法》及相关配套制度在药品注册管理制度、流程、药品的全生命周期质量管理、药品上市许可持有人制度、专利链接制度等方面都做出了重大调整和创新,在全面贯彻落实“四个最严”要求下,提升了我国药品研发的效率,加速了国内药品向国际化拓展的进程,加强了药品知识产权的保障,促进了药品研发和生产资源的流转,为我国制药行业的健康发展奠定了坚实的制度基础。
The newly-revised Drug Administration Law and its relevant supporting regulations made major adjustments and innovative changes on the management procedures of drug registration and supervision, lifecycle quality management of drugs, marketing authorization holder system and drug patent linkage system. Under the “four strictest” requirements for drug quality management, the Law has improved the efficiency of China’s drug R&D, accelerated the advance of domestic drugs to the global stage, strengthened the protection of intellectual property rights relevant to drugs, promoted the circulation of drug R&D and production resources, and laid a solid institutional foundation for the healthy development of China’s pharmaceutical industry.
关键词Key words
新药品管理法;新药;仿制药;研发;生产
newly-revised drug administration law; new drugs; generic drugs; research & development; manufacturing
《药品管理法》自1984 年9月20 日发布以来,经过2013 年和2015 年2 次修正,2001 年和2019 年2 次修订,现行《药品管理法》于2019 年12 月1 日正式实施。新修订《药品管理法》基于激励国内药品研发、规范制药行业生产经营活动、强化药品监督管理、促进药品产业健康发展、维护公众健康权益的顶层设计。以符合我国国情为前提,借鉴国内外药品管理监督有益经验,在理念和体制、制度和系统、机制和方式、战略和文化等方面改革创新,坚持科学发展[1]。2020年和2021 年,新药品管理法对制药行业带来的影响已逐步显现。药品注册管理制度的优化,激发了国内药品研发热情;药品质量管理制度的强化,提升了药品质量水平;专利链接制度体系的建立,加强了药品知识产权的保护;上市许可制度的确立,促进了药品研发、生产资源的流转。本文将以制药行业从业者视角,总结归纳新修订《药品管理法》实施以来国内医药行业的变化。
一、药品注册监管的变化
2019 年12 月新修订《药品管理法》实施以来,国家药品监督管理局着力完善药品审评审批工作制度,新修订了《药品注册管理办法》,推进了审评审批制度改革,完成了审评审批流程的全面优化。以临床价值为导向,满足日益提升的医疗需求为目标,构建了新的药品注册分类;优化了监管流程、提高监管效率;明确了审评时限;将审评、核查及检验工作由原来的“串行”改变为“并行”;设立突破性治疗、附条件批准、优先审评审批、特别审批4 个快速通道;确定了原料药、药用辅料和药包材(即原辅包)的关联审评政策。使国内药品注册管理,从制度到流程都有了较大的突破。
(一)国内药品研发的变化
由图1 所示,自新修订《药品管理法》实施以来,国内药品注册申请数量大幅提升,2020 年药品注册申请共计10247 件,较上年同比增长26.87%。2021 年药品注册申请达到11497 件,较上年同比增长12.20% ;其中,2021 年化药8147 件, 较上年同比增长3.38% ;2021 年生物制品2006 件,较上年同比增长7.39% ;中药申请大幅上升,达到1317 件, 较上年同比增长179.03%。
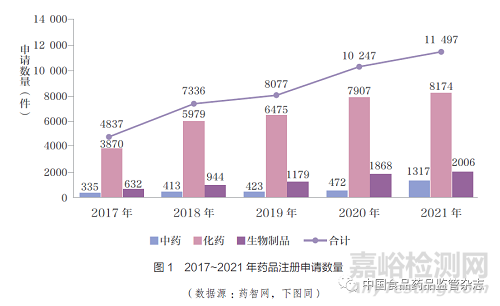
如图2 所示,近2 年新药申报数量也迅速增加,2020 年较2019 年化药新药申报数量增加47.85% ;2021 年较2020 年增长58.09% ;其中化药1 类同比增长56.10% ;化药2 类(改良型新药)同比增长65.77%。
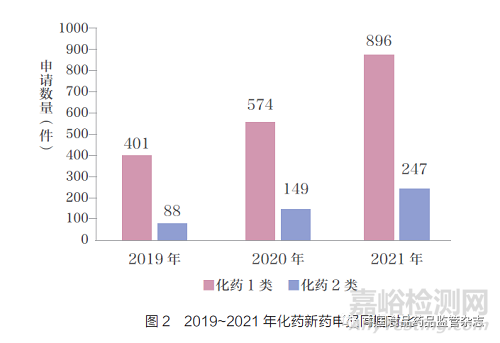
如图3 所示,在生物药方面,2019~2021 年新生物制品申报数量迅速增长,2021 年较2019 年总体增幅达到236.54%,其中治疗用生物制品从2019 年的194件增长到2021 年的675 件。预防用生物制品数量变化较小。
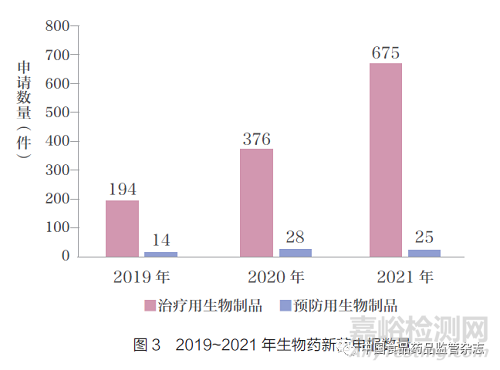
如图4 所示,2021 年中药新药申报数量增长较快,新中药申报数量从2019 年的18 件增长到2021 年的62 件,改良型中药2021 年无新申报。
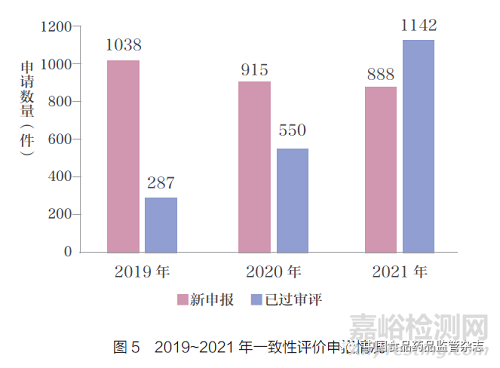
如图5 所示,在一致性评价方面:2021 年一致性评价申报共计888 件,较2020 年的915 件略有下降。而随着一致性评价审评的加速,2021 年共计已过审评1142件, 相较于2020 年(550 件)和2019 年(287 件)大幅增加。
综上,在新修订《药品管理法》及配套政策的支持下,我国医药行业研发活力较好,在新药申报方面2020 年至2021 年均有大幅增加。
(二)“加速通道”制度
新修订《药品管理法》及《药品注册管理办法》在加速审批相关制度上作了较大的调整,鼓励研究和创新药、鼓励儿童用药的研制和创新、鼓励短缺药品的研制和生产,新增了突破性治疗药物和附条件批准两条程序,调整了优先纳入范围,明确了审评时限。突出了鼓励“具有临床价值”药品开发,早期介入,有效沟通,并确定在提出上市许可申请时可一并提出优先审评申请。
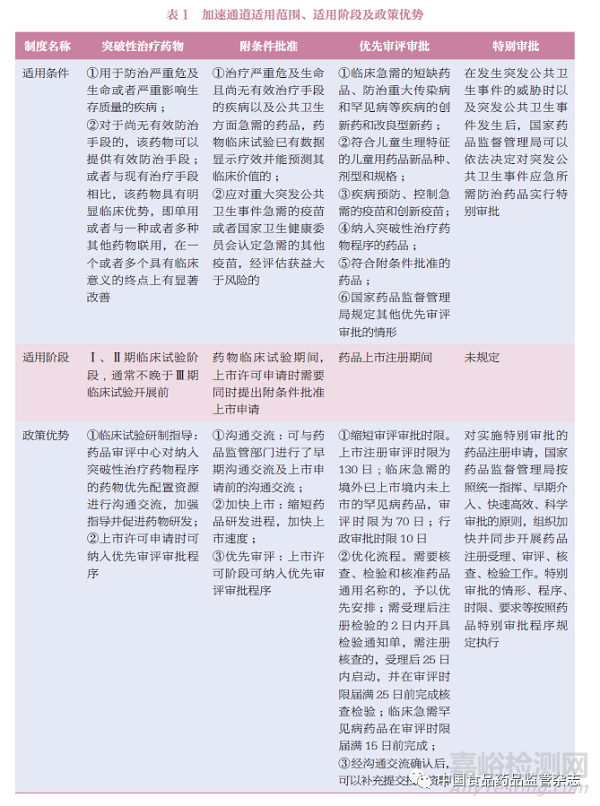
加速通道包括突破性治疗药物、附条件批准、优先审评审批、特别审批。新的优先审评审批程序中将审评时限确定为130 日,将临床急需、境外已上市而境内未上市罕见病药品的审评时限确定为70 日;在审评阶段,可在沟通交流确认后补充提交技术资料,核查、检验以及通用名称核准等程序一并予以优先安排。见表1。
我国的药品加速通道立足于国情,又参考国际药品快速注册制度经验。在国际上,美国FDA最早在1983 年开始使用孤儿药法案,用来鼓励、加速罕见病的上市,在1988 年启动了快速通道机制以加速药物临床评估进程,而后在1992 年和2012 年分别设立了加速审批路径和突破性疗法认证。美国FDA 在罕见病、严重疾病的治疗药物和治疗方法的快速审批方面都具有成熟的经验。在国内,2005 年发布实施了针对突发公共卫生事件应急所需药品加快审评审批的程序,后续在2016 年对优先审评审批的范围、程序和工作要求作出规定, 而2017 年和2018 年分别对创新药、临床急需的境外药品的优先审评审批制定了相应的工作程序;在新修订的《药品管理法》中积极借鉴了美国FDA 突破性疗法认证程序,延续了我国原有的药品特别审批程序,优化调整了优先审评审批程序。与美国FDA 的快速通道政策相比,主要有4 点差异:①在通道数量上:我国的药品加快上市注册程序未单独设立快速通道。②在纳入程序上:我国附条件批准程序中监管部门会对试验设计及所附条件等进行技术指导,而美国FDA 的加速审批无资格认定程序。③在提出时间上:我国突破性治疗是在药品临床试验获得批准后提出,而美国FDA的突破性疗法可在提交临床试验申请的同时提交,也可在之后提交。④在适用范围上:在各项加速审评通道的适用范围上有差异,使其更贴近我国的临床需求、患者自身特点、流行病学情况以及药品监管的需要[2]。
在快速通道下审评审批的药品上市周期大幅缩减, 首个国产口服酪氨酸激酶(BTK) 抑制剂药物奥布替尼片, 是用于治疗复发/ 难治性套细胞淋巴瘤(MCL)、慢性淋巴细胞白血病/小淋巴细胞淋巴瘤(CLL/SLL)的1 类新药,在2020 年3 月提交新药注册申请,在优先审评通道下历时9 个月,即在2020 年12 月获批生产。同样,用于治疗携带FLT3 突变的复发性或难治性急性髓性白血病(AML)成人患者的富马酸吉瑞替尼片在2020年4 月提交新药申请(NDA)进口申请,在优先审评通道下,也是历时9 个月在2021 年1 月获批进口。由此可见,加速通道的政策大大提高了重大疾病、罕见病药物的上市速度。
(三)临床试验指南逐步与国际接轨
新修订《药品管理法》及《新药品注册管理办法》实施后,国家药品监督管理局陆续出台了数十个关于药物和医疗器械的临床试验的指导原则和规范。涵盖了化药、生物药、中药和医疗器械等产品类别,涉及肿瘤、皮肤和尿路感染、脂质代谢紊乱等多种疾病,覆盖到临床设计及临床试验管理等多个环节。此外,还出台了多个生物类似物品种临床试验指导原则。
随着国内的临床试验指南及指导原则逐步与国际接轨,我国制药企业在药品临床试验及注册技术要求的理解方面逐步贴近国际化要求,提高国内药品向国际拓展的成功率,为中国制药企业向跨国药企迈进打下坚实的基础[3]。使得国内外临床试验数据能够无障碍桥接,为国外已获批的新药快速进入中国打下基础。而国际型药企的研发模式和研发经验在国内可得到有效的桥接,进一步强化创新合作,并有意愿将更多的新药开发工作在我国进行,更有利于创新药在国内的落地。
知屋漏者在宇下,作为制药行业从业者,笔者在药品研发过程中仍会遇到如有些国外已上市的非处方药品无法以国内现行药品注册路径进行申报的情况,也希望未来药品监管部门能够推进相应技术指导原则的落实,以期将更多的具有确切疗效的非处方药品引入国内。
(四)审评审批信息公开管理
新修订《药品管理法》要求批准上市药品的审评结论和依据应当依法公开,接受社会监督。国家药品监督管理局药品审评中心在2020 年12 月31 日发布的《药品审评审批信息公开管理办法》,已于2021 年6 月1 日开始实施。该办法明确了受理信息、审评审批信息、审评审批结果信息、临床试验基本信息、技术审评报告、药品说明书、原辅包信息以及申请人反馈,以及审评过程中汇总的共性问题等的公开范围。此项政策的出台提高了药品审评审批工作的透明度,使得药品监管部门对药品注册申请人的引导作用得到提高,并可及时消除因技术评价标准不明确、指导原则未规定而产生的共性问题、疑难问题的不利影响。
(五)逐步构建“三结合”中药审评证据体系
2021 年6 月24 日国家药品监督管理局发布了《关于实施中国药品监管科学行动计划第二批重点项目的通知》[4],将加快推进中医药理论、人用经验、临床试验三结合审评证据体系的构建纳入重点项目,从根本上解决中药临床疗效评价的难题,从而大幅提升中药审评审批速度,在这样政策利好的情况下,2021 年度中药新药申报数量大幅提升。但由于国内的中药研发力度积弱已久,目前中药新药的总体申报数量与化药和生物药相比仍有较大的差距,未来增长空间较大。如果未来中药新药申报数量达到目前化药和生物药水平,以目前中药审评审批来看,将会是一个挑战。
二、具有“ 中国特色”的药品质量管理
新修订《药品管理法》在立法过程中全面贯彻了药品安全“四个最严”要求。积极借鉴当今国内外药品监管的有益经验,药品风险管理的最新实践成果,并立足国情,充分考虑当前我国药品产业特点、药品监管体制、社会治理文化等因素。
(一)动态药品生产质量管理规范(GMP) 监管取代GMP认证制度
在新修订《药品管理法》实施后,国家药品监督管理局加大了对药品研发、生产、经营全过程的监管力度,以动态监管替代既往的GMP/ 药品经营质量管理规范(GSP)等相关认证制度,药品监管工作重点由事前许可向事中、事后监管转变[5]。除许可检查、常规检查和有因检查等监督检查工作外,针对原辅包的延伸检查也被纳入省级药监部门的检查范围,并引入了告诫信制度作为监管新措施[6]。制药企业在药品监管部门动态监管的影响下,逐步从硬件型验证向药品生产全过程的质量控制转变。
(二)中国特色的药物警戒制度
新修订《药品管理法》提出:“国家建立药物警戒制度,对药品不良反应及其他与用药有关的有害反应进行监测、识别、评估和控制。”这标志着我国正式引入了药物警戒制度,明确了在药品上市许可持有人(MAH)制度下,持有人在不良反应监测报告与处理、风险识别与评估、上市后安全性研究、药品安全风险控制等方面所担负的法律责任。
2021 年12 月1 日,国家药品监督管理局发布的《药物警戒质量管理规范》正式实施[7],在借鉴国际先进药物警戒管理经验同时,创新提出了具有中国特色的中药、民族药管理要求。同时《药物警戒检查指导原则(征求意见稿)》[8] 也于当日公开征求意见,作为《药物警戒质量管理规范》的重要支撑。该指导原则的出台,对药品上市许可人在药物警戒体系的构建及完善起到了重要指导作用。
由于我国的药物警戒制度刚刚确立,与国际先进的药物警戒体系相比,我国的药物警戒制度处在起步阶段。因此,对国外先进理念和措施的借鉴必不可少[4]。在最近2 年中,国内制药企业积极与药品监管部门、行业内先驱者等沟通交流,逐步建立符合中国国情的药物警戒体系[9]。
(三)最严格的药品质量责任管理
新修订《药品管理法》首次在信用管理方面引入了联合惩戒制度,国家药品监管部门负责建立药品安全信用档案,包含记录许可颁发、日常监督检查结果、违法行为查处等信息,对有不良信用记录的增加监督检查频次,并可按照国家规定实施联合惩戒[10-11]。
新修订《药品管理法》还引入了管理者处罚到人的机制,将企业的主体责任与管理者担责相结合,综合运用了警告、罚款、没收、停产停业、吊销许可证、禁止从业等多种处罚方式,大幅提高处罚力度,并单独设定了刑事处罚。
三、专利链接制度体系的建立
在国内创新药和仿制药产业加速发展的形势下,如何做好原研药品和仿制药品生产企业之间的利益关系的平衡、如何在鼓励创新的前提下避免对专利权的滥用、如何完成符合中国国情的专利链接制度的构建,一直是国家药品监督管理局与国家知识产权局对国家药品监管和专利保护政策研究的重要议题。药品专利链接相关法律法规不断出台,中国的专利链接制度逐步形成体系。
2021 年6 月1 日,《专利法》修订案[12] 正式施行。修改后的《专利法》规定了在专利授权过程中的不合理延迟,以及在新药上市审评审批占用时间的专利期限补偿,并对药品专利纠纷早期解决机制进行规定,在药品行政审批与专利保护之间进行了有效链接。
专利链接相关法规实施后,使原研药品专利的作用在现有基础上得到进一步的发挥,原研药企业也会选择合理的专利策略进一步延长产品的生命周期。同时,仿制药立项之初就会对原研药的专利布局和稳定性进行深入研究,以保证项目顺利推进。能否通过专利挑战获得首仿药的市场独占期,以及能否将首仿药的市场独占期与集采窗口期有效结合,可能会成为未来化药仿制药项目成败的关键。
四、药品上市许可的变化
新修订《药品管理法》确立了药品上市许可持有人制度,对药品上市许可持有人的权利、义务、责任进行了明确划分,从原来只有药品生产企业可以进行药品注册申请,转变为取得药品注册证书的企业或者药品研制机构等均可成为药品上市许可持有人,其他的相关政策也激发了制药行业药品开发的积极性。
(一)激活批准文号内涵价值
新修订《药品管理法》明确了经国务院药品监督管理部门批准,药品上市许可持有人可以转让药品上市许可(药品批准文号),改变了以往只能技术转让,无法进行批准文号转让的情况。这促进了药品上市许可在市场上流转,持有人为其付出的时间、资金、技术、智慧,乃至药品监管部门的审评审批工作成果等也可以通过市场流转实现其价值。
(二)上市许可、生产许可分离,激活研发能动性
新修订《药品管理法》上市许可持有人制度解除了生产许可和上市许可之间的绑定,改变了以往国内研发机构在不进行投资建厂的条件下, 无法将新药研发成果转化, 无法形成良性循环的矛盾。使得药品研发机构(CDMO)将药品生产委托给药品合同生产企业(CMO)或具有闲置产能的药品生产企业,从而集中资金、技术和人力进行药品的持续研究和新药品的开发。
(三)优化国内医药产业结构
新修订《药品管理法》上市许可持有人制度实施前,药品批文无法流转,新药研发难以落地,市场空间大的药品批文成为各药品生产企业竞逐利润的基础,可能会有盲目扩大生产以降低生产成本提高产品竞争力的情况发生,造成生产资源重复或空置浪费[13-15]。在上市许可持有人制度落实后,允许CMO 或具有闲置生产资源的药品生产企业进行药品生产承接,进而达成国内药品生产资源的合理调配;使得研发型企业和生产型企业能够各司其职,促进国内药品产业结构的调整和升级。
目前,国内已有多家基于药品上市许可持有人政策的信息平台上线,以找品种、找厂家、找服务为基础, 为合同研究组织(CRO) 公司、CMO 公司、研发机构、生产企业进行服务推动资源整合和规模化集约化生产。
五、总结和展望
新修订《药品管理法》实施的2 年以来,从各方面已经表现出国内药政改革的决心,相关配套规章制度也陆续出台,优化了国内药品监管体系,激发了国内药品研发活力,强化了药品知识产权管理,促进了国内医药产业优化整合,提升了药品全生命周期质量管理理念。随着新修订《药品管理法》的配套措施的不断完善,我国的药事活动正在逐步走向现代化和国际化。可以预见的是,随着新规章、新制度的不断实施,新临床指南、新指导原则的持续引领,我国的制药行业将很快走进新的时代。

来源:中国食品药品监管杂志


