您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2020-03-06 14:55
摘要:
伴随大量创新及仿制药面世而来的是对遗传毒性杂质加强监管的迫切需求。一系列与国际接轨的指南性文件的出台,弥补了我国杂质监管的空白,但难点尚存。传统评价方法具有局限性,新方法与传统方法间缺乏一致性比较,药物特定合成路线实际产生的大量含警示结构杂质缺乏毒理学评价数据支持,毒性预测软件的预测效力不足等。本文就国内外杂质遗传毒性杂质监管科学现状、遗传毒性杂质控制策略、杂质遗传毒性评价方法进行论述,并提出充分结合计算机毒理学、毒性评价方法和符合我国国情的监管限度制定三个维度开展杂质监管工作。
关键词:杂质 遗传毒性 评价方法 警示结构 细菌回复突变试验 Pig-a基因突变试验
2018年7月6日,浙江华海药业公布其抗高血压药物缬沙坦的原料药中发现遗传毒性杂质(Genotoxicity Impurity)N-亚硝基二甲胺(N-nitrosodimethylamine,NDMA)的含量超过限值。NDMA作为N-亚硝胺类化合物,被国际癌症研究机构(International Agency for Research on Cancer,IARC)列为2A类致癌物,少量摄取即存在诱发肿瘤的风险。2018年7月23日,华海药业迅速完成国内缬沙坦原料药的召回。2018年7月29日,原国家食品药品监督管理总局新闻发言人对华海药业NDMA杂质事件作情况说明。2018年8月17日,国家药典委员会对缬沙坦的国家标准进行修订并指出必须对生产工艺进行评估以确定形成N-亚硝基二甲胺的可能性。此事在国内引起轩然大波,并再次唤醒公众对遗传毒性杂质监管的重视。在事件的持续发酵过程中,华海药业股价连续跌停,年度净利润下滑78.4%,累计计提损失超过4亿元。华海药业并非第一家涉及遗传毒性杂质管控事故的厂家,也绝非最后一个。2007年,欧洲药品管理局(European Medicines Agency,EMA)发现抗艾滋病药物Viracept中含有的遗传毒性杂质甲磺酸乙酯(Ethyl Methanesulfonate,EMS)超出限定值,从欧洲市场召回[1]。2019年6月,美国麦克劳德制药(Macleods Pharmaceuticals)生产的2批次氯沙坦钾片和30批次的氯沙坦钾/氢氯噻嗪片也因检出超量NDMA而自愿从市场召回[2]。一系列药品召回事件造成恶劣社会影响,并不断敲响药物杂质毒性监管的警钟。
1 国内外遗传毒性杂质监管现状
药品在生产合成和贮运过程中可能引入对人体有害的杂质,其中一部分具有遗传毒性风险的成为遗传毒性杂质。后者在很低浓度下即可诱导基因突变和染色体损伤,甚至导致癌症并危及生命[3]。遗传毒性杂质本质上是药物生产或保存过程中难以避免,甚至不可避免的产物。制药企业在药物研发过程中,通常需花费大量时间和财力来验证合成过程中不存在有遗传毒性风险的产物。遗传毒性杂质的出现也对监管机构提出了新的要求和挑战。然而,遗传毒性杂质的监管起步较晚,自2000年以后才陆续受到各国的重视。近年来,欧盟、美国药品监管机构相继发布了遗传毒性和致癌性杂质的指导原则。如EMA人用药委员会安全工作组(Committees for Human Medicinal Products,CHMP)于2004年发布《遗传毒性杂质限度指南》并首次提出毒理学相关阈值(Threshold of Toxicological Concern,TTC)限度的概念[4];美国食品药品监督管理局(Food and Drug Administration,FDA)于2006年发布《原料药和成品药中遗传毒性和致癌性杂质推荐方法》并提出使用决策树(Decision Tree)法对杂质风险进行评估[5];人用药品注册技术要求国际协调会(International Council for Harmonization of Technical Requirement for Pharmaceuticals for Human Use,ICH)在2017年发布的ICH M7指导原则中为遗传毒性杂质的确认、研究和控制方法提供了指导性的建议和技术要求[6]。ICH M7的颁布具有里程碑式的意义,它将取代EMA遗传毒性杂质限度指南和美国FDA遗传毒性杂质指南草案,成为今后国际上广泛通行的遗传毒性杂质控制指导性文件。原国家食品药品监督管理局于2017年成为ICH正式成员,我国相关标准和指导原则已与国际接轨。《中华人民共和国药典》 (以下简称《中国药典》)2015年版四部中已收载了《药品杂质分析指导原则》,国家药典委员会于2019年及时发布了《遗传毒性杂质控制指导原则》 (征求意见稿) [7],当前,国家药品监督管理局正在推进ICH M7在我国的落地与转化。一系列指南性文件的出台弥补了我国与先进国家和组织之间的贸易与技术壁垒。
2 遗传毒性杂质控制策略
致癌性风险在药物研发阶段即受到高度重视,通常会在研发早期开展基因突变遗传毒性标准组合试验对其致癌风险进行评估,而有潜在风险的化合物则及早停止研发。杂质因其特性有可能无法从药物中去除,故根据人体暴露情况设置定量限成为可行的监管方略。EMA于2006年首先在遗传毒性杂质监管中引入化学物质慢性毒性研究的毒理学相关阈值概念,在700余种致癌物分析基础上,提出以1.5μg·d-1为有潜在遗传毒性风险的化合物的TTC[8]。该暴露水平下,人体终生服用该物质的患癌可能性为十万分之一。美国药品研究和制造商协会(The Pharmaceutical Research and Manufacturers of America,PhRMA)进一步对建立暴露时间与TTC之间的关联,建立“分期TTC [9]”。此外,PhRMA率先提出了杂质遗传毒性五分类的概念,并以构效关系(Structure-activity Relationship,SAR)为分类依据,根据警示结构预测其诱导人体肿瘤的风险[9]。从而将化合物结构作为碱基插入导致的突变和肿瘤形成的重要决策基础,而评估碱基突变风险的试验方法也在分类中有重要权重。“分期TTC”和五分类决策树的概念对全球药品杂质监管影响深远,美国FDA的《遗传毒性杂质指南草案:原料药和成品药中遗传毒性和致癌性杂质》、ICH M7及我国的《遗传毒性杂质控制指导原则》相继沿袭,是当前杂质监管领域的主要思路。值得注意的是“分期TCC”的具体限量在不同指南及法规中有所不同,如表 1所示,我国则以ICH M7为制定监管标准的重要依据。本文以ICH M7为蓝本就杂质监管要求作简要介绍。
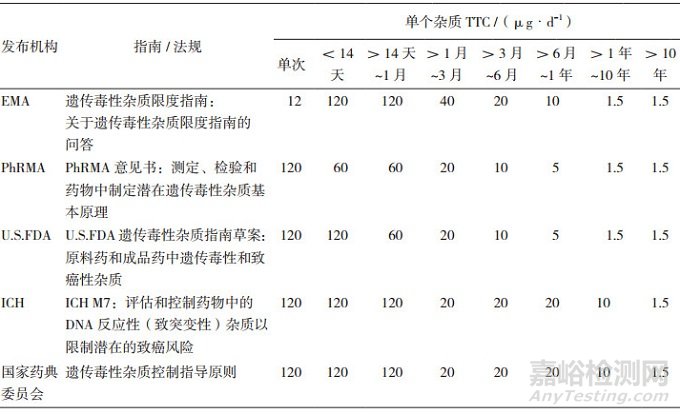
表 1 不同指南及法规关于单个杂质服用时间与TTC限量对比
ICH M7主要适用于DNA反应活性杂质,即在含量很低时便存在致碱基突变及诱发人体肿瘤的风险。遗传毒性杂质毒性的确认及定量构效关系(Quantitative Structure-activity Relationship,QSAR)数据库的构建均以毒性研究数据为基础。根据致突变和致癌风险危害程度,ICH M7将所有杂质归为5类并提出相关控制策略[6]。1类杂质:已知具有致突变性的致癌物。对于该类杂质,首先考虑去除杂质,如果无法去除,则应建立基于未观察到作用水平(No Observed Effect Level,NOEL)致癌性数据的限度数值。2类杂质:致癌性未知的已知致突变物。如存在阈值相关机制,以每日允许暴露量(Permitted Daily Exposure,PDE)求限度值;如无阈值相关机制的证据,根据TTC确立限度。3类杂质:含警示结构、与原料药结构无关且无致突变性数据的物质。根据TTC确定限度值,或根据致突变性试验评价结果归为2类杂质或5类杂质并作相应控制。4类杂质:含警示结构、与原料药或原料药相关物质的警示结构相同,且经测试认为无致突变性的物质;按5类杂质(普通杂质)控制。5类杂质:无警示结构,或有充分的数据证明警示结构无致突变性或致癌性的物质。按普通杂质控制。
判断某个杂质是否属于遗传毒性杂质时,首先应收集其相关细菌回复突变试验、动物和人体致癌性数据。部分公开致癌性数据库[10]如表 2所示。

表 2 常用化合物致癌性数据库信息
2.1 已有致癌性或致突变性试验数据的杂质控制
杂质具有已知致突变性或致癌性数据归为1类致癌物,根据半数致癌剂量(MedianToxic Dose,TD50)线性外推法对其进行控制,即以诱导50%肿瘤发生率的剂量的五万分之一为摄入量,可将肿瘤的发生风险控制于十万分之一。致癌性强度数据库(Carcinogenicity Potency Database,CPDB)中已明确列出1547种致癌物的TD50值供公开查询。在测定TD50时,应以单一动物种属和单一性别特定器官的最低TD50为准。无致癌性试验数据的物质,归为第2类或第3类的杂质,服用时间超过10年的药物,单一杂质的摄入量为1.5 μg·d-1,可以应用TTC作为大多数毒性化合物作为可接受摄入量的默认值[11]。如原料药质量标准中存在两个2类或3类杂质,应制定各自限度值;如果含3种或更多的2类或3类杂质,可按多个杂质的总可被接受摄入量(服用时间超过10年的药物杂质5 μg·d-1)计算(如表 2)。值得注意的是,1类杂质和制剂中形成的降解产物应单独控制,不计入2类和3类杂质的总限度值。黄曲霉素类物质、N-亚硝基化合物和偶氮类等强致突变性化合物属于关注队列(Cohort of Concern,COC) [6],TTC法对该类化合物不适用,其TD50应低于1.25 mg·(kg·d) -1。某些DNA反应性化合物可能受到某种代谢或调控通路影响,其实际阈值或摄入量与致癌性呈非线性关系,如乙基甲烷磺酸盐、过氧化氢、苯胺类化合物等,可使用NOEL或PDE[计算公式:(NOEL×体质量)/(动物与人用剂量差异系数×不同人体之间差异×研究时间系数×严重情况系数×观察到反应的最低剂量)]确定限量值(如表 3所示) [6]。
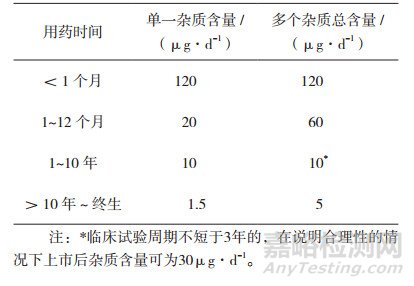
表 3 ICH M7中TTC法控制遗传毒性杂质限度[6]
2.2 无已知致突变性及致癌性试验数据的杂质控制
绝大多数杂质缺乏充分的遗传毒性和致癌性研究数据,往往难以归类。遗传毒性的“警示结构”指杂质中的某些特殊基团或亚单位可与遗传物质发生化学反应,从而导致基因突变或染色体重排/断裂,具有潜在致癌风险。而产生遗传毒性致癌性的化合物,从作用机制上分析均具有DNA反应活性,通过与遗传物质发生化学键合引起遗传信息改变产生致癌性,而基因突变往往是遗传毒性致癌性作用的起始环节[12-13]。Ashby和Benigni等根据高DNA反应活性可以认为是碱基突变及肿瘤形成最初阶段的原理,归纳并提出了18种化学结构的“警示结构(Alert Structure)”模型,包括酰卤、烷基或苯基磺酸酯、烷基或苯基磷酸酯、N-羟甲基衍生物、单卤代烯烃、硫芥、氮芥等[14],如图 1所示。随后,在国际上提出采用“警示结构”作为区分普通杂质和遗传毒性杂质的重要依据的概念。这一概念已广泛应用于遗传毒性和致癌性预测软件,也成为遗传毒性警示结构以及QSAR模型的基础。对于存在“警示结构”的杂质进行QSAR预测和体内外相关研究,或将其含量控制在一定范围之内。当前已有多种计算机软件可对化合物的遗传毒性进行预测,如基于文献报道和专家知识的预测软件Toxtree(Joint Research Centre of European Chemical Bureau)、Derek Nexus(Lhasa Limited)和基于统计学的预测软件如Sarah Nexus(Lhasa Limited)和CASE Ultra(MultiCase Inc.)等。这些预测决策树或根据既往Ames试验或微核试验等遗传毒性试验数据,或根据警示结构的遗传毒性阳性可能性来生成判断结果。
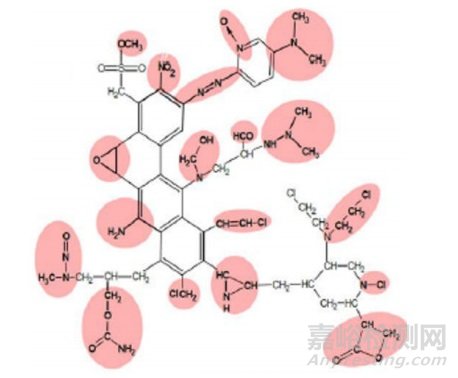
图 1 含有多种“警示结构”的超级致癌物的提出[12]
然而,现有数据库在预测效力上存在一定局限性。新药研发催生大量新型有效药物成分(Act ive Pharmaceutical Ingredient,API)和新型合成路线的诞生,现有的数据库信息与浩如烟海的杂质种类相比乃九牛之一毛。一项针对十余家跨国药企百余种合成路线产生的杂质的研究中共发现600多种警示结构,其中烷化剂、芳香胺、酰胺类及芳香硝基衍生物类警示结构占总警示结构的61.3%[15]。值得关注的是含酰氯衍生物、烷基醛及硼酸类警示结构的化合物在该研究中占总警示结构的5.8%、4.7%和3.5%,但在已知致癌信息数据库中仅占0.5%、0%和0%。故上述警示结构的化合物的致癌性有必要进行试验研究。此外,含有“警示结构”的化合物并非一定具有遗传毒性。以常用解热镇痛药对乙酰氨基酚(4-Acetamidophenol/N-acetylp-aminophenol,APAP)为例,2011年国家药品抽检的APAP药品中含有多种苯酚类杂质,基于决策树的毒性预测平台Toxtree预测其中绝大多数含有遗传毒性致癌性警示结构、多数含Ames致突变性试验阳性警示结构和体内微核试验阳性警示结构,如表 4所示。然而,实际临床及动物长期研究数据并不提示APAP存在致癌性。再以ICH M7推荐使用的毒理学数据库Derek Nexus为例,其对截至2005年已有206种化合物的预测结果的一致性为86%,灵敏性为73%;而对2005-2011年间新增的化合物的预测度的一致性为89%,灵敏度降低至50%[16-17]。比如,Derek Nexus对4-氯乙酰基乙酰苯胺的致突变性预测结果为阴性,然而在多个实验室共同开展Ames试验联合验证工作中均得到明确阳性结果[18]。预测灵敏度的降低提示存在致突变风险的化合物无法通过软件预测的方法获知,监管工作中仅凭借构效分析软件预测结果仍然存在风险。此外,某些创新药的化学结构可能难以通过数据库进行预测。无论基于经验或统计学的数据库的构建,其预测结果的准确度都极大程度上依赖于真实的试验数据。可见开展试验研究对杂质毒性评价具有重要价值。
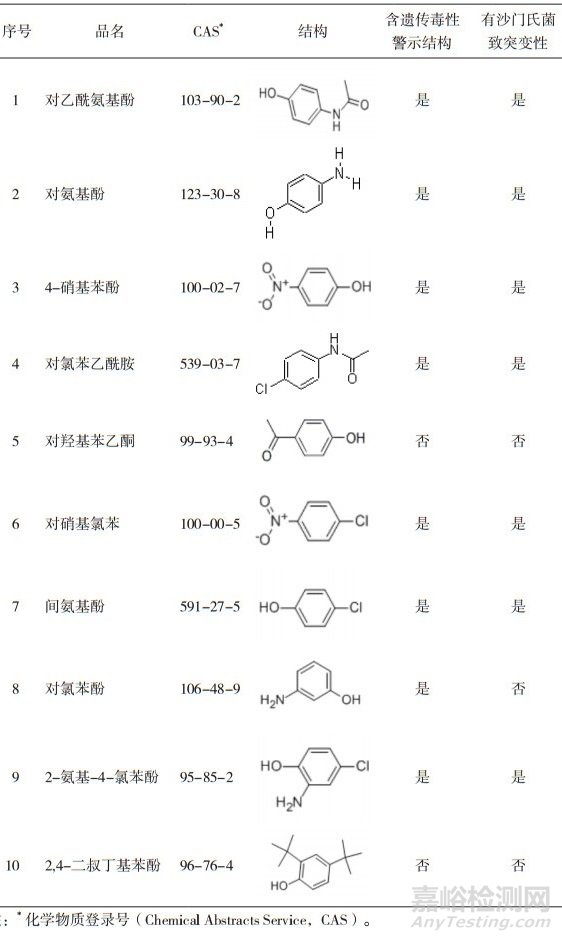
表 4 Toxtree对乙酰氨基酚及其杂质遗传毒性预测结果
3 药物杂质致突变性评价方法
由遗传毒性发生的机理可知,基因突变是肿瘤发生的分子基础。以碱基突变为评价终点的Ames试验和体内Pig-a基因突变试验,均作为杂质遗传毒性风险评价的首选试验方法[6],其试验数据已获得FDA认可。评价受试物与DNA作用风险的彗星试验结果对杂质遗传毒性评价也有一定参考。杂质毒性评价的另一大难题,是可合成或获取的杂质量难以满足试验需要,而ICH M7要求尽量使用杂质纯品开展评价;此外,单一化合物可能有10~20余种杂质。受试物需求量少、试验周期短的快速评价方法才能满足大量杂质遗传毒性评价的迫切需求,故有必要优化和改进传统遗传毒性评价方法。
3.1 Ames试验
细菌回复突变试验(Bacterial Reverse Mutation Test,Ames),1970年由Bruce. N. Ames[19]使用组氨酸营养缺陷型鼠伤寒沙门氏菌株(S.Typhimurium)建立,故命名为Ames试验。该试验利用缺乏合成组氨酸能力的细菌在可诱导碱基突变的化合物的作用下可通过发生回复突变,从而自行合成组氨酸来形成大量肉眼可见的菌落的原理,通过菌落计数来评价受试物的碱基突变能力。该方法是当前所有遗传毒性评价方法中对致癌物预测度最高的试验,Ames试验对大鼠致癌物和恒河猴致癌物的检出率分别为69.0%和87.5%,也是化合物QSAR构效关系建立的遗传毒性筛选数据库的重要基础[20]。
随着分子生物技术发展的飞跃,Ames作为一项经典的致突变性筛选方法经历了一系列改良,比如微孔板、液态培养法或引入含荧光报告基因等[21-22]。ICH M7认可基于标准平皿的Ames试验数据。但某些杂质合成困难,基于微孔板的Ames试验,如Mini-Ames(6孔板)或Micro-Ames(24孔板)在不改动传统Ames试验原理的条件下可减少杂质的用量,极具应用潜力。OECD正在对Ames试验指导原则进行修订,并于2018年在国际范围内收集Mini-Ames、Micro-Ames和Ames波动试验的背景数据,试图形成标准化的操作流程。然而非标准平皿的Ames试验各家实验室操作流程差异很大,较难形成统一操作标准,可能对审评工作造成一定难度。开展相关试验方法与传统试验方法结果的一致性比较则有助于数据互认和评价效率。笔者完成的Ames与Mini-Ames试验中阴性与阳性对照组回复突变菌落数比较如表 5所示。
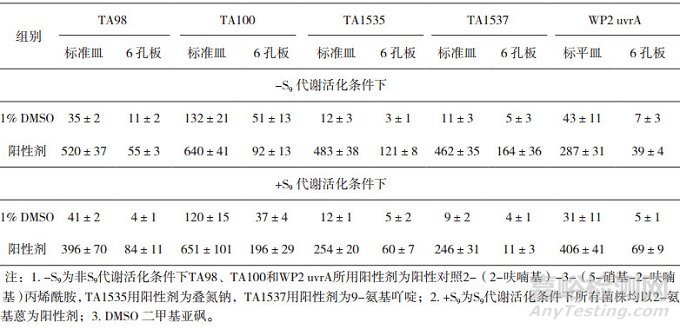
表 5 标准平皿Ames与Mini-Ames试验回复突变菌落数(个/皿,x±s)
3.2 Pig-a基因突变试验
体内Pig-a基因突变试验是近年来在药物安全性评价领域日益受到重视的一项药物致突变性评价方法。Pig-a基因位于X染色体,该基因突变可导致糖化磷脂酰肌醇(Glycosylphos-phatidyl Inositol,GPI)合成障碍,从而使相关细胞GPI锚蛋白(如CD48、CD55、CD59等)缺失。因GPI锚蛋白在不同物种间的生物合成高度保守,且在各类组织细胞及各类成熟红细胞中均有表达,故试验可使用正常动物外周血作为试验系统[23]。Araten等[24]在1999年首次提出Pig-a基因突变试验可用药物基因突变风险评价。Dertinger等[25]进一步将高通量免疫磁性筛选技术引入Pig-a基因突变试验,有效提高了统计功效。该方法当前已完成多中心联合验证[25-26],OECD拟于2020年正式发布相关指导原则。当Ames试验结果不明确时,体内Pig-a基因突变试验可作为后续选择的试验方法,其结果已获得美国FDA认可。体内Pig-a基因突变试验的主要优势在于可检测弱致突变剂体内蓄积后的致突变风险,并获得受试物对啮齿类动物致癌风险的相关数据。但其缺点也较为突出,即受试物需求量较大,试验周期长,对于合成量较少的杂质来说存在一定困难,且不适合大量杂质毒性筛选。但体内Pig-a基因突变试验尚未经过国内联合验证及推广,实际上具有相关试验能力和经验的国内安全性评价中心很少。
近年来,国内外陆续开展基于TK6、MCL-5以及L5178Y等哺乳动物细胞系的体外Pig-a基因突变试验建立与验证工作[27-29]。体外Pig-a基因突变试验的优点是使用的受试物用量少、试验周期短(一般受试物处理8天后检测)、不使用动物和适于进行机制研究。但是,在安全评价领域正式使用之前,在细胞选择和标准化试验方法的验证等方面还需要开展大量工作。体内及体外Pig-a基因突变试验优劣对比详见表 6。
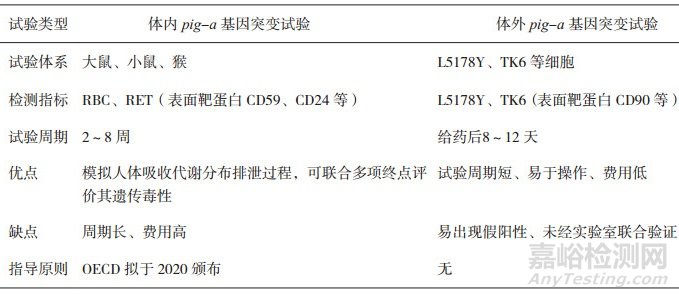
表 6 体内及体外Pig-a基因突变试验比较
3.3 彗星试验
Ostling和Johansson[30]于1984年建立彗星实验(Comet Assay),也称为单细胞凝胶电泳(Single Cell Gel Electrophoresis,SCGE),可在单细胞水平上检测DNA链断裂程度,从而评价受试物是否与DNA相互作用。该方法利用损伤的DNA与完整的DNA在电泳中迁移速率差异,对存在DNA损伤的细胞(形成彗星样结构)进行区分。因DNA断裂多在给药后短期内出现,该方法可灵敏地检出短时间内出现的DNA损伤。但DNA单链断裂可自行修复,损伤不一定遗传给子代细胞。故彗星试验的结果可作为参考,用于判断受试物与DNA碱基作用的可能性,其结果对杂质的遗传毒性有一定预测作用。国际肝细胞彗星试验实验室间联合验证于2015年完成并发表[31],OECD于2014年颁布体内彗星试验指导原则[32]。
4 小结
综上所述,随着大量创新药及仿制药的出现,迫切需要加强遗传毒性杂质的监管。我国已陆续出台了一系列与国际接轨的指南性文件以支持杂质的毒性评估,但实际工作中难点尚存。如传统方法在杂质评价中存在局限性,指南性文件对杂质的遗传毒性评价要求不明确,缺乏新方法与传统方法之间的一致性比较,药物合成路线实际产生的大量含警示结构杂质无毒理学研究数据支持,基于构效分析的毒性预测软件的预测效力不足等。此外,通过对国家药品抽检数据库与欧洲药典分析比对,我国国抽药品实际检出杂质与欧洲药典中所列出的杂质类型存在较大差异,其原因可能与药物合成加工方式不同有关。我国药典中对药品可能存在的杂质和具体定量限要求缺乏详细明确的界定,而欧洲药典中的杂质毒性及定量限的信息又难以借鉴。上述因素对监管人员在药物化学、遗传毒性评价等多领域的知识储备提出了新的挑战。遗传毒性杂质的监管不是纸上谈兵,应充分结合计算机毒理学、毒性评价方法和制定符合我国国情的监管限度值,从三个维度切实做好杂质的控制,从而提高药物的安全性,保障人民用药安全。
参考文献
[1] Pozniak A, Muller L, Salgo M, et al. Elevated Ethyl Methanesulfonate (EMS) in Nelfinavir Mesylate (Viracept®, Roche):Overview[J]. AIDS Res Ther, 2009, 6: 18. DOI:10.1186/1742-6405-6-18
[2] U.S. FOOD & DRUG ADMINISTRATION. FDA Updates and Press Announcements on Angiotensin Ⅱ Receptor Blocker (ARB) Recalls (Valsartan, Losartan, and Irbesartan)[S]. 2019.
[3] Bercu JP, Dobo KL, Gocke E, et al. Overview of Genotoxic Impurities in Pharmaceutical Development[J]. Int J Toxicol, 2009, 28(6): 468-478. DOI:10.1177/1091581809349195
[4] EUROPEAN MEDICINES AGENCY. Guideline on the Limits of Genotoxic Impurities[S]. 2006.
[5] U.S. FOOD & DRUG ADMINISTRATION. Draft Guidance for Industry on Genotoxic and Carcinogenic Impurities in Drug Substances and Products: Recommended Approaches; Availability[S]. 2008.
[6] EUROPEAN MEDICINES AGENCY. ICH Guideline M7(R1) on Assessment and Control of DNA Reactive (mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk Step 5[S]. 2018.
[7] 国家药典委员会.遗传毒性杂质控制指导原则(第一次征求意见稿)[S]. 2019.
[8] Kroes R, Renwick A G, Cheeseman M, et al. Structurebased Shresholds of Toxicological Concern (TTC):Guidance for Application to Substances Present at Low Levels in the Diet[J]. Food Chem Toxicol, 2004, 42(1): 65-83. DOI:10.1016/j.fct.2003.08.006
[9] CHMP Safety Working Party. Question & Answers on the CHMP Guideline on the Limits of Genotoxic Impurities[S]. 2009.
[10] Amberg A, Beilke L, Bercu J, et al. Principles and Procedures for Implementation of ICH M7 Recommended (Q) SAR Analyses[J]. Regul Toxicol Pharmacol, 2016, 77: 13-24. DOI:10.1016/j.yrtph.2016.02.004
[11] Munro IC, Ford RA, Kennepohl E, et al. Correlation of Structural Class with No Observed Effect Levels:A Proposal for Establishing a Threshold of Concern[J]. Food Chem Toxicol, 1996, 34(9): 829-867. DOI:10.1016/S0278-6915(96)00049-X
[12] Ashby J. Fundamental Structural Alerts to Potential Carcinogenicity or Noncarcinogenicity[J]. 1985, 7(6): 919-921.
[13] 马磊, 马玉楠, 陈震, 等. 遗传毒性杂质的警示结构[J]. 中国新药杂志, 2014, 23(18): 2106-2111.
[14] Ashby J, Tennant RW. Chemical Structure, Salmonella Mutagenicity and Extent of Carcinogenicity as Indicators of Genotoxic Carcinogenesis among 222 Chemicals Tested in Rodents by the U.S. NCI/NTP[J]. Mutat Res, 1988, 204(1): 17-115.
[15] Galloway SM, Reddy MV, Mcgettigan K, et al. Potentially Mutagenic Impurities:Analysis of Structural Classes and Carcinogenic Potencies of Chemical Intermediates in Pharmaceutical Syntheses Supports Alternative Methods to the Default TTC for Calculating Safe Levels of Impurities[J]. Regul Toxicol Pharmacol, 2013, 66(3): 326-335. DOI:10.1016/j.yrtph.2013.05.005
[16] Hayashi M, Kamata E, Hirose A, et al. In Silico Assessment of Chemical Mutagenesis in Comparison with Results of Salmonellamicrosome Assay on 909 Chemicals[J]. Mutat Res, 2005, 588(2): 129-135. DOI:10.1016/j.mrgentox.2005.09.009
[17] Hillebrecht A, Muster W, Brigo A, et al. Comparative Evaluation of in Silico Systems for Ames Test Mutagenicity Prediction:Scopeand Limitations[J]. Chem Res Toxicol, 2011, 24(6): 843-54. DOI:10.1021/tx2000398
[18] Dunkel VC, Zeiger E, Brusick D, et al. Reproducibility of Microbial Mutagenicity Assays:Ⅱ. Testing of Carcinogens and Noncarcinogens in SalmonellaTyphimurium and Escherichia Coli[J]. Environ Mutagen, 1985, 7 Suppl 5: 1-248.
[19] Mortelmans K, Zeiger E. The Ames Salmonella/Microsome Mutagenicity Assay[J]. Mutat Res, 2000, 455(1): 29-60.
[20] 于仲波, 吴南翔, 金锋, 等. 1485种化学物致突变试验和致癌试验结果一致性比较[J]. 毒理学杂志, 2007(4): 320-320.
[21] Zwart N, Lamoree M, Houtman C, et al. Development of a Luminescent MutagenicityTest for High-throughput Screening of Aquatic Samples[J]. Toxicol In Vitro, 2018, 46: 350-360. DOI:10.1016/j.tiv.2017.09.005
[22] 文海若, 宋捷, 鄂蕊, 等. 微孔板与标准平皿Ames试验比较研究[J]. 药物评价研究, 2019, 42(5): 81-86.
[23] Peruzzi B, Araten DJ, Notaro R, et al. The use of PIG-A as a Sentinel Gene for the Study of the Somatic Mutation Rate and of Mutagenic Agents in Vivo[J]. Mutat Res Rev Mutat Res, 2010, 705(1): 3-10. DOI:10.1016/j.mrrev.2009.12.004
[24] Kawagoe K, Takeda J, Endo Y, et al. Molecular Cloning of Murine Pig-a, a Gene for GPI-Anchor Biosynthesis, and Demonstration of Interspecies Conservation of Its Structure, Function, and Genetic Locus[J]. Genomics, 1994, 23(3): 566-574. DOI:10.1006/geno.1994.1544
[25] Dertinger SD, Phonethepswath S, Weller P, et al. Interlaboratory Pig-a Gene Mutation Assay Trial:Studies of 1, 3-propane Sultone with Immunomagnetic Enrichment of Mutant Erythrocytes[J]. Environ Mol Mutagen, 2011, 52(9): 748-755. DOI:10.1002/em.20671
[26] Gollapudi BB, Lynch AM, Heflich RH, et al. The in Vivo Pig-a Assay:A Report of the International Workshop On Genotoxicity Testing (IWGT) Workgroup[J]. Mutat Res Genet Toxicol Environ Mutagen, 2015, 783: 23-35. DOI:10.1016/j.mrgentox.2014.09.007
[27] Kruger CT, Fischer BM, Armant O. The in Vitro PIG-A Gene MutationAssay:Glycosylphosphatidylinositol (GPI)-related Genotype-to-phenotype Relationship in TK6 Cells[J]. Arch Toxicol, 2016, 90(7): 1729-1736. DOI:10.1007/s00204-016-1707-x
[28] David R, Talbot E, Allen B, et al. The Development of an in Vitro Pig-a Assay in L5178Y Cells[J]. Arch Toxicol, 2018, 92(4): 1609-1623. DOI:10.1007/s00204-018-2157-4
[29] 李若婉, 周长慧, 黄鹏程, 等. 基于TK6细胞的体外PIG-A基因突变检测方法的建立[J]. 癌变·畸变·突变, 2019, 31(3): 242-248.
[30] Collins AR. Measuring Oxidative Damage to DNA and its Repair with the Comet Assay[J]. Biochim Biophys Acta, 2014, 1840(2): 794-800. DOI:10.1016/j.bbagen.2013.04.022
[31] Uno Y, Kojima H, Hayashi M. The JaCVAM-organized International Validation Study of the in Vivo Rodent Alkaline Comet Assay[J]. Mutat Res, 2015, 786-788: 45-76. DOI:10.1016/j.mrgentox.2015.04.010
[32] OECD. Guidelines for the Testing of Chemicals No. 489: In Vivo Mammalian Alkaline Comet Assay[S]. 2014.
作者:文海若, 闫明, 王亚楠 1, 王翀 3, 耿兴超 1, 朱炯 3, 张河战, 王雪
1. 中国食品药品检定研究院 国家药物安全评价监测中心, 药物非临床安全评价研究北京市重点实验室, 北京 100176;
2. 中国药科大学, 南京 210009;
3. 中国食品药品检定研究院技术监督中心, 北京 102629

来源:xml-data


