您当前的位置:检测资讯 > 法规标准
嘉峪检测网 2019-04-22 22:50
作者:张丹(航天中心医院临床药理室)
编辑:春雪
作者简洁:张丹,女,工程师,硕士,主要从事药物分析和药动学研究。
[摘要]生物等效性试验是药品研发和评价的一个重要内容。生物等效性试验首选药动学研究的方法.药动学研究则必然涉及到生物分析,因此生物分析对于生物等效性试验具有举足轻重的作用,其结果的可靠性将直接影响生物等效性试验的质量。但我国目前却没有完整、独立的生物分析方面的指导原则。基于上述原因,我们根据国内外的相关指导原则,并尽可能结合我们的实际工作,探讨生物等效性试验中生物分析需注意的关键问题。
许多注册类别的药品(如创新药、药物新剂型、仿制药、部分补充申请的药品)研发和评价都涉及到生物等效性试验,其中药物新剂型、仿制药、补充申请的药品在完成生物等效性试验后即可能被批准上市,而不需要进行其他临床研究。另外,目前我国的药品质量一致性评价也会更多地涉及到生物等效性试验,足见生物等效性试验在药品研发和评价中的重要性。生物等效性研究的方法包括体内和体外的方法,按方法的优先考虑程度从高到低排列:药动学研究方法、药效学研究方法、临床比较试验方法、体外研究方法。首选药动学研究的方法必然涉及到生物分析。药动学方法是采用人体生物利用度比较研究的方法,通过测量不同时间点的生物样本(如全血、血浆、血清或尿液)中药物浓度,得出与吸收程度和速度有关的药动学参数.经统计学比较后判断制剂间是否具有生物等效性。因此生物分析对于生物等效性试验具有举足轻重的作用,其结果的可靠性将直接影响生物等效性试验的质量。
欧洲药品管理局(EuropeanMedicinesAgency。EMA)和美国食品和药物管理局(USFoodandDrugAdministration.FDA)均有生物分析方面的指导原则,我国国家食品药品监督管理总局(ChinaFoodandDurgAdministration,CFDA)虽有指导原则涉及相关内容,但却不完整、不独立。目前,中国生物分析协会(ChinaBioanalysisFourm。CBF)正在讨论中国药典2015年版的生物分析指导原则草案(《生物样品定量分析方法指导原则(草案)》),旨在提供建设性的意见。该草案则主要参考了EMA的“Guidelineonbioanalyticalmethodvalidation”(2011)、FDA的“GuidanceforIndustry,BioanalyticalMethod Validation”(2001)、中国药典2010年版的《药物制剂人体生物利用度和生物等效性试验指导原则》中“生物样品分析方法的基本要求”、以及CFDA相关指导原则中涉及生物样品定量分析方法的内容(2005)。EMA的“Guidelineonbioanalyticalmethodvalidation”(2011)中共8次提及“生物等效性试验”,EMA、FDA除上述指导原则外.另有与生物等效性试验中生物分析相关的指导原则,再次体现了生物分析在生物等效性试验中的重要地位。因此,我们根据国内外的相关指导原则,并尽可能结合我们的实际工作,针对生物等效性试验中生物分析的分析物确定及分析工作开展的注意事项等关键问题,谈谈我们的一些认识和做法。
生物等效性试验生物分析的分析物确定
有别于一般的药动学研究,生物等效性试验的生物分析,确定分析物的原则是:该分析物的药动学参数应足以反映制剂间吸收速度和吸收程度的差异。这些参数包括常见的药物浓度-时间曲线下面积(AUC包括AUC0-t、AUC0-∞ 、AUCss部分曲线下面积AUCpantial)、峰浓度(pmax)、达峰时间(tmax),也有一些仍在探讨中的参数,比如Pmax/AUC、平均驻留时间(MRT)、控释制剂平台时间(t75%max血药浓度维持在最大浓度75%以上的时间)等。根据这一原则,我们从以下三个方面进行探讨。
1母药和代谢物
1.1测定母药
由于母药的JD比代谢物的JD一更能反映制剂间吸收速度的差异,因此无论母药有无活性,在生物分析技术条件允许的情况下,通常应测定母药。
氯吡格雷作为前体药物,其本身并无活性,在体内约85%的氯吡格雷形成无活性的羧酸代谢物氯吡格雷酸,少部分形成不稳定的中间活性代谢物(硫醇代谢物)抑制血小板的聚集,而血浆中氯吡格雷的浓度很低,在生物分析技术条件不足的时期不易被检测。活性代谢物在血浆中的浓度也较低.且稳定性很差.常规分析方法难以对其准确测定.在样本采集同时进行衍生化虽可解决稳定性的问题,对活性代谢物的专利保护导致其商业化对照品的缺乏,也制约了该分析技术的应用。因此,在早期的氯吡格雷口服制剂生物等效性试验中,大多测定氯吡格雷酸并以其进行生物等效性评价或增大给药剂量后测定氯吡格雷并进行生物等效性评价。但随着分析技术不断的发展.已能准确可靠地测定口服氯吡格雷后血浆中母药氯吡格雷的浓度,在生物等效性试验时则应测定母药氯吡格雷棚。当然,当分析技术发展到可以准确可靠地测定口服氯吡格雷后血浆中活性代谢物的浓度.并得以广泛应用时.也许生物等效性试验的分析物将拓展到其母药和活性代谢物均应测定。
1.2测定活性代谢物
当母药是无活性的前体药物,且药物浓度一时间曲线由于血药浓度过低或消除太快而不完整时,应测定其活性代谢物。非诺贝特在吸收的同时被组织和血浆中的酯酶迅速、完全地水解为活性代谢物非诺贝特酸,而在血浆中检测不到母药非诺贝特,因此只能通过检测非诺贝特酸的浓度来反映非诺贝特吸收的速度和程度。
1.3测定母药和主要活性代谢物
当母药和代谢、物均有活性,尤其是活性代谢物在体内的暴露量较大时,一般应测定母药和主要活性代谢物,以母药进行生物等效性统计和评价,以主要活性代谢物进一步支持生物等效性评价的结论。对于代谢物,只需提供其浓度、药动学参数,以及AUC和p的几何均数、几何均数比值(geometricmeanratios,GMRs)。
氯雷他定在体内的主要活性代谢物是去羧乙氧基氯雷他定(地氯雷他定),两者均应测定。人体内氯雷他定的消除半衰期(t1/2)约为9h,而地氯雷他定的t1/2约为27h,在生物等效性试验的生物样本采集点的设计方面就需要两者兼顾。阿托伐他汀在体内的主要活性代谢物是邻羟基阿托伐他汀和对羟基阿托伐他汀,3个化合物均应测定。但需要注意的是,FDA建议的生物等效性试验用药剂量为80mg,而在我国的常用起始剂量为10mg,即便试验采用20mg的给药剂量,体内对羟基阿托伐他汀的浓度也很低,可能不易测定,因此目前已报道的阿托伐他汀钙的生物等效性试验大多只测定了阿托伐他汀和邻羟基阿托伐他汀。
阿苯达唑在体内首先代谢成活性代谢物阿苯达唑亚砜(代谢率高达98%以上),最终代谢成无活性的阿苯达唑砜。生物等效性试验应测定阿苯达唑和阿苯达唑亚砜。特殊的是.首选母药进行生物等效性统计和评价。但如果确实有实验来证明母药不能被准确、精密的测定时,则可以选择活性代谢物阿苯达唑亚砜进行生物等效性统计和评价。
2手性药物——消旋体和对映体
对于手性药物的生物等效性试验,我们应考虑测定消旋体还是对映体,或者说是选择非手性的分析方法还是手性的分析方法。
EMA一般可接受非手性方法.但在以下情况时应采用手性方法:
(1)若两个对映体具有不同的药动学性质、明显不同的药效学性质、且系统暴露比值受吸收速度的差异所影响.应采用手性方法测定所有对映体单体,当上述对映体的性质未知时也应采用手性方法;
(2)若一个对映体有活性,而另一个对映体无活性或活性较低,采用手性方法测定,且只需要以活性对映体进行生物等效性统计和评价。
FDA认为对于生物利用度试验.有必要测定对映体单体。而对于生物等效性试验一般推荐用非手性方法测定消旋体。但若两个对映体具有不同的药动学、药效学性质.体内暴露较低的对映体对疗效和安全性起决定性作用,且至少一个对映体具有非线性吸收性质(表现为对映体在体内的浓度比值随吸收速度的变化而变化)时,应采用手性方法,并分别以对映体单体进行生物等效性统计和评价。
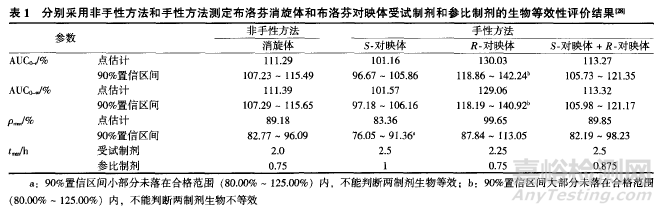
非甾体抗炎药布洛芬是一个手性药物.其右旋布洛芬(S-对映体)和旋布洛芬(R-对映体)具有不同的药动学、药效学性质,且对映体在体内的浓度比值受吸收速度的影响。TORRADO等进行了布洛芬混悬剂的生物等效性试验,分别采用非手性方法和手性方法测定了布洛芬消旋体和布洛芬的两个对映体单体,并进行了生物等效性评价,结果列于表1。依据FDA指导原则中规定的评价标准,当采用非手性方法测定布洛芬消旋体时,结论是受试制剂和参比制剂生物等效:然而,采用手性方法测定布洛芬对映体时,结论是对于有效的一对映体,不能判断两制剂生物等效。受试制剂的生物利用偏低;对于无效的、有副作用的一对映体,不能判断两制剂生物不等效,受试制剂的生物利用度偏高;但是将两个对映体加起来,结论又是两制剂生物等效。可以看出,采用非手性方法和手性方法可能会得到截然不同的评价结果,应慎重选择非手性方法代替手性方法。
结合我们完成的一些手性药物制剂生物等效性试验的生物分析工作,我们认为:
(1)严格来讲,手性化合物的对映体本质上是不同的化合物,虽然它们除旋光性不同,其他物理性质、化学性质均相同,但多数具有不同的生理活性.因此手性药物的生物分析理应采用手性方法。
(2)在不增加生物等效性试验结果风险的前提下,某些情况还是可以用非手性方法代替手性方法的.最主要的两种情况是:
①对映体在吸收方面无立体选择性,即在跨膜转运、首关效应无立体选择性;
②无活性或活性较低的对映体在体内暴露量较小。
3多组分药品
针对多组分药品的生物等效性试验分析物的确定.FDA指出应该测定足以反映吸收速度和吸收程度的标志化合物,应综合考虑:各组分在制剂中含量的多少、在体内浓度的高低、以及生物活性的大小。对于不同的多组分药品,应具体情况具体分析。目前,我国对于西药复方制剂的要求是各组分均应测定。需要强调的是,多组分药品生物等效性试验分析物确定时仍需要考虑“1.1母药和代谢物”、“1.2手性药物——消旋体和对映体”中提及的问题。另外,在进行复方制剂生物等效性试验的生物分析时,还需要关注并考察各分析物的测定是否会受到其他分析物(共存组分)的影响。
苯磺酸氨氯地平/阿托伐他汀钙片有效成分为苯磺酸氨氯地平和阿托伐他汀钙,根据相关指导原则和要求.进行生物等效性试验时两个成分均应测定,且均应进行生物等效性统计和评价,还需要测定阿托伐他汀的主要活性代谢物邻羟基阿托伐他汀和对羟基阿托伐他汀,并提供相应的数据进一步支持生物等效性评价的结论。
复方氨酚烷胺口服制剂有效药物成分为对乙酰氨基酚、盐酸金刚烷胺、咖啡因、马来酸氯苯那敏和人工牛黄。其中,人工牛黄是由牛胆粉、胆酸、猪去氧胆酸、牛磺酸、胆红素、胆固醇、微量元素等加工制成的,其中胆酸和胆红素为主要成分,中国药典规定人工牛黄中胆酸和胆红素含量分别不得少于13.0%和0.63%。目前尚无天然牛黄或人工牛黄在人体内药动学研究的报道。在我们完成的一项复方氨酚烷胺口服制剂的生物等效性试验中。受试制剂和参比制剂中人工牛黄的用药剂量均为12mg,若以胆酸和胆红素含量分别为13.O%和0.63%计算,则含胆酸1.56mg、胆红素75.6ug。胆红素的用药量很小,即使口服后完全被吸收,也不足以影响受试者体内胆红素的正常水平,而对受试者用药前后血浆中胆酸浓度的测定结果也表明人体内内源性胆酸的浓度较高,且个体内浓度水平相对较稳定。用药前后受试者体内的胆酸浓度无明显改变.但个体间差异较大。因此,我们参考了FDA关于多组分药品的生物等效性试验分析物的确定原则,测定了对乙酰氨基酚、金刚烷胺、咖啡因、氯苯那敏的血药浓度并以其主要药动学参数进行了生物等效性评价。而未将人工牛黄主要成分胆酸和胆红素作为评价的指标。或许可以参考FDA的这个原则进行我国中成药的生物等效性研究,提高我国对中药的监管水平,以确保中药的质量。
生物等效性试验生物分析中的注意事项
分析测定应在一个合适的范围内准确、精密地测定未知生物样本。由于生物等效性试验的特殊性,生物分析的结果可能会对生物等效性试验中方差分析的误差来源及误差大小产生影响。针对这一问题,我们根据相关指导原则。结合我们的工作,从以下几个方面谈谈生物等效性试验中生物分析时需关注的内容。
1分析批
EMA和FDA关于分析批的内容、合格标准在细节上虽略有不同,但在原则上基本一致。
两者均要求在方法确证之后方可进行未知生物样本的分析测定,一个完整的分析批至少应包括:用于建立标准曲线的空白样本、零点(zero)样本(空白样本添加工作浓度的内标)、标准曲线,未知生物样本.以及随行的均匀分布在未知生物样本测定过程中的、数量不低于该分析批未知生物样本总数5%、至少3个浓度水平且每一浓度水平至少2样本的质量控制(Qc)样本。而FDA还指出应在分析批之前进行系统适应性(systemsuitability)的考察。
两者均要求至少3/4的标准曲线样本的测定浓度与理论浓度之间相对误差(RE)不应超出±15%的范围,定量下限(LLOQ)可放宽至±20%,且合格的浓度点总数应不少于6个,采用合格的浓度点进行线性回归分析.线性回归方程的相关系数r应大于0.99。标准曲线测定结果不符合上述要求时,该标准曲线作废。不可用于当批未知生物样本的分析测定,应重新建立可满足要求的标准曲线。此外,EMA还指出若LLOQ样本不合格,则以合格的最低浓度点作为LLOQ,若定量上限(ULOQ)样本不合格,则以合格的最高浓度点作为ULOQ,修正后的标准曲线线性范围应能覆盖全部的QC浓度水平.若同一分析批建立2条标准曲线、且仅1个LLOQ或ULOQ样本不合格,标准曲线的线性范围不需要修正。
EMA要求至少2/3、且同一浓度水平至少1/2的QC样本的测定浓度与理论浓度的RE应不超过±15%,LLOQ浓度点可放宽至±20%,FDA的要求基本相同,不同的是不合格的Qc样本不能全部出现在同一浓度水平中。QC样本测定结果不符合上述要求时,该分析批未知生物样本的测试结果作废,需要重新处理并测定。此外,两者也均要求除考察每个分析批的QC样本的准确度与精密度之外.还应考察所有可接受分析批各浓度水平的QC样本的总体准确度与精密度,计算时应包括这些分析批中不合格的QC样本。合格标准仍然是RE不超过±15%,相对标准偏差(RSD)不超过15%。EMA还特别强调Qc样本的总体准确度与精密度不符合要求时。应另外进行考察来解释导致这种偏离的原因,而对于生物等效性试验,Qc样本的总体准确度与精密度不符合要求很可能会导致整体数据作废。
我们一直关注并依循EMA和FDA指导原则的最新要求进行生物分析。但结合我们在生物等效性试验中生物分析的经验,对某些细节的要求,尤其是它们的不同之处,有一些自己的认识:
(1)关于系统适应性的考察,我们遵循FDA的原则,在分析批开始之前.应至少重复6次进样测定同一个标准样本(所含溶剂、生物基质等最好与分析批中所测样本一致),计算相关分析物及内标响应值的RSD,RSD不超过15%(标准样本为生物样本处理所得时)可证明仪器运行状态适合该分析批样本的测定,如RSD不合格,则应采取适当措施改良仪器运行状态使其适合样本的测定。
(2)考虑到消除相浓度逐渐接近LLOQ或大于LLOQ的那一个最低浓度点,若LLOQ不合格而以满足要求的最低浓度点作为LLOQ,则很可能导致消除相原本可测定的浓度点严重缺失,并影响t1/2、AU/0-∞等参数的计算以及生物等效性评价的结果,而EMA也提出在某分析批LLOQ不合格而导致某些样本浓度不可测时,还需要在LLOQ合格的分析批中复测这些样本;另外,ULOQ作为标准曲线的最高浓度点。其RE与低浓度点相比理应较小,方法建立及验证时未出现由于仪器响应超载
(overload)而导致的ULOQ不合格,却在未知生物样本测定时出现ULOQ不合格的现象。则很可能是与其他一些错误操作有关。因此我们认为标准曲线的LLOQ和ULOQ必须满足要求,不满足要求时不可对标准曲线的线性范围进行修正,该标准曲线应按作废处理,采取适当措施寻找原因加以避免,并重新建立可满足要求的标准曲线。
(3)考虑到生物等效性试验每个分析批测定的样本量可能较大.尤其是在方法通量高的情况下,分析批中的QC样本总数可能较大,我们认为现有原则关于同一浓度水平最多允许不合格QC样本数量的要求都略有不妥。若根据FDA的要求,可能会出现不合格QC样本的总体数量增加但分析批却反而从不合格变为合格的情况,可见FDA的要求不甚合理,而我国CFDA要求不合格的Qc样本“不能出现在同一浓度质控样品中”又过于严苛,相比之下EMA的要求更为合理.但也略微宽松,允许同一浓度水平中出现较多不合格的0C样本可能会导致位于该浓度水平附近的未知生物样本的测定结果不准确的风险增加。因此.我们采用的标准是:同一浓度水平至多允许1/3的QC样本的测定结果不合格。
上述要求是确保准确、精密测定未知生物样本的最基本的要求,应严格地遵循以确保生物等效性试验的统计结果的质量。
2处理批
由于生物等效性试验每个分析批的生物样本量较大,通常我们无法同时处理一个分析批所有受试者的样本,而需要分批处理。为了减少分析测定引起的不必要的个体内差异。同一名受试者的所有未知生物样本应同时处理.并随行处理低、中、高浓度的QC样本。当然,如果样本中分析物的稳定性比较差,则需要综合考虑分析物可稳定的时长、试验的周期数及清洗期时长、样本分析测定所需时长等因素,安排分析批和处理批。
3盲测
为避免主观意识对分析测定结果的影响,EMA要求生物分析应处于盲测的状态。结合我国开展生物等效性试验的一些现状,我们认为盲测是很有必要的。从事生物等效性试验生物分析的人员不仅不应知道所测样本是受试者服用受试制剂(testformulation,T)还是参比制剂(referenceformulation,R)之后所采集的,甚至连分组情况也不应知道。完成分析测定之后,在分析测定报告中只需要列出所测样本的编号及对应的测定浓度,分析测定工作完成、数据核对、锁定之后方可揭盲。
4复测
在生物分析的过程中,可能会出现某分析批首次测定数据作废,仪器进样、操作的失误,测定浓度超过ULOQ、色谱行为差、内标响应异常等情况。而需要复测。复测包括重新进样(reinjection)、重新处理再测定(reprocessingandreanalysis)。EMA和FDA均要求无论因何原因进行何种复测,在未知生物样本分析测定工作开始之前,就应该在试验方案、或研究计划、或相关标准操作规程(standardoperatingprocedures,SOPs)中规定复测的理由、复测的方法、以及测定值的取舍标准,在报告中也应提供原始测定值、复测的理由、复测值、最终接受值、接受的标准、以及复测样本量占总样本量的比例。但在复测的理由上.EMA和FDA可接受的情况略有不同,我们认为:
(1)样本处理后的样液体积足以重新进样,且方法确证已证明重新进样的重现性良好、分析物于处理后在进样器中的稳定性良好,由于
仪器原因导致进样失败或者个别样本在进样后色谱行为差(如峰形差、保留时间改变)时,才可以通过重新进样来复测样本。
(2)在以下情况时,需要重新处理再测定——重新进样结果仍然异常;由于仪器原因导致进样失败(包括单个的样本、以及整批未能继续进样的样本)或者个别样本在进样后色谱行为差,但样液体积不足以重新进样,或可重新进样的时间已超出分析物于处理后在进样器中的稳定时长;由于QC样本不满足要求导致整个分析批作废;测定浓度高于ULOQ;内标响应值明显高于或低于当前分析批标准样本(标准曲线样本及QC样本)内标平均响应值,但需要事先量化这种“内标响应值异常”:在用药前采集的样本中检测到分析物响应值高于LLOQ,但需要结合合理的测定误差范围和分析物性质进行判断,如是否为内源性物质。
(3)对于生物等效性试验而言,一般不允许因出现不符合药动学趋势的高浓度值或低浓度值而复测,因为这样的复测很可能会导致生物等效性评价结果的偏倚。即便复测,也仅是为了探索导致异常的原因.以避免类似的问题再次发生。
5已测样本再分析(incurredsamplesreanalysis.ISR)
由于在方法验证时使用的标准样本和QC样本可能无法模拟试验采集到的实际生物样本。例如,蛋白结合、已知和未知代谢物的逆向转化、样本均一性、合并用药所引起的差异.可能导致实际样本在保存和处理过程中分析物浓度的变化,而使测定结果的准确度和精密度受到影响。因此,应考虑通过ISR.即在一定时间周期内重新分析已测的生物样本。来评价实际生物样本测定结果是否准确可靠、可重现。
FDA并无关于ISR的要求,但EMA要求所有关键的生物等效性试验的生物分析均需要进行ISR,并提供了详细的操作要求及合格标准:测定的未知生物样本总数少于1000时应对1O%的样本进行ISR,测定的未知生物样本总数超过1000时应对5%的样本进行ISR;应选择接近丁pmax以及在消除相的样本:两次测定浓度值的差异应不超过20%,且至少67%的ISR样本的测定结果应符合此要求。
我们认为,鉴于ISR的主要目的是评价实际样本分析测定结果的是否准确可靠、可重现,其反映的是一系列因素的综合结果,这其中包括分析方法、仪器状态、样本的采集、保存、处理等各方面的因素,因此即便是采用相同的分析方法测定相同的分析物。只要是不同的试验(不属于同一次样本采集、同一次生物分析),每个生物等效性试验的生物分析都应该进行ISR;ISR应尽早进行,且在分析物的稳定时长内完成;样本总数为1000时应对l0%的样本(100个)进行ISR,样本总数超过1000时应对5%的样本进行ISR,并且ISR的样本量应不少于样本总数为1000时ISR的样本量(100个),也就是说样本总数为1000~2000时,ISR的样本量为100个;ISR样本的选择除了应选择不同受试者的接近于pmax以及在消除相的样本外,还需注意不应选择超过ULOQ、已经复测或需要复测的样本。
结语
目前我国生物等效性试验生物分析的现状迫切需要规范化。因此我们根据国内外的相关指导原则.在总结实际工作经验的基础上,探讨了生物等效性试验中生物分析需注意的关键问题,希望能对其规范化提供可参考的价值。同时,我们也期待有关指导原则的早日出台,这将有利于提高我国生物等效性试验的质量,提高我国药品研发的水平。

来源:AnyTesting


