您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2019-02-15 09:38
目的:依据《中国药典》2015年版, 对抽检的41批医院制剂进行微生物限度检查和质量分析。
方法:检验方法为制剂生产医院提供, 并根据药典要求, 对每批样品进行控制菌检查时均加入阳性对照。
结果:抽检的41批医院制剂样品微生物限度检查, 37批次检验结果均符合规定, 1批次检验结果不符合规定, 3批样品由于医院提供的微生物限度检查的方法依据为2010版《中国药典》, 不符合现行药典要求, 故而未做检验。
结论:医院制剂微生物限度检查方面存在诸多问题, 应引起各方重视。
医疗机构制剂(以下简称医院制剂), 是指医疗机构根据本单位临床需要经批准而配制、自用的固定处方制剂。医院制剂作为药品的一种特殊补充形式, 长期以来, 因其便捷、有效等特点在临床诊疗服务中发挥着重要作用。医院制剂质量标准是反映医院制剂质量特性的技术参数、指标, 执行标准是控制医院制剂质量的有效手段。制剂质量直接关系到用药者的安全和疗效。而微生物限度检查在制剂质量控制方面起着重要的质量评估作用, 其准确与否直接关系到患者用药的安全性和有效性。医院制剂的微生物污染可以造成制剂的变质、失效或产生毒性。本次抽检的样品, 检验依据均为《天津市医疗机构制剂规范》2016年版。该规范中相关的微生物限度检查项目规定均是指向《中国药典》2015年版四部通则。因此, 本次抽样检查, 所参考的检验方法及标准规定均为《中国药典》 2015年版四部通则1105、1106和1107。本文旨在对天津市部分医院制剂进行微生物限度检查, 对制剂微生物污染情况及存在的问题和原因进行分析, 为规范天津市医疗机构质量监督和制剂管理、完善医疗机构制剂质量标准提供参考。
1 仪器与材料
1.1 仪器
BINDER BD240 恒温培养箱(德国BINDER); BINDER KB720恒温培养箱(德国BINDER); SANYO MIR-254恒温培养箱(日本三洋株式会社); SANYO MIR-554恒温培养箱(日本三洋株式会社); SG-403A TX-INT生物安全柜(美国Baker); SQP精密电子天平(德国赛多利斯); Lab dancer漩涡混合器(德国IKA); Yamato810C高压灭菌锅(日本Yamato株式会社); 薄膜过滤器(英国Whatman公司); SHAKE4000-8CE摇床(美国Thermo公司); YamatoADS161SMU超净工作台(日本Yamato株式会社)。
1.2 试验菌种
大肠埃希菌(Escherichia coli)[CMCC(B)44 102]、乙型副伤寒沙门菌(Salmonella paratyphi B )[CMCC (B)50094]、金黄色葡萄球菌(Staphylococcus aureus)[CMCC (B)26 003]、铜绿假单胞菌(Pseudomonas aeruginosa)[CMCC (B) 10104], 白色念珠菌(Candida albicans) [CMCC (F) 98001], 均购自中国食品药品检定研究院。
1.3 培养基及稀释剂
胰酪大豆胨琼脂培养基TSA (批号160214)、胰酪大豆胨液体培养基TSB (批号160316)、沙氏葡萄糖琼脂培养基SDA (批号170316)、麦康凯液体培养基(批号160229)、麦康凯琼脂培养基(批号160405)、甘露醇氯化钠琼脂培养基(批号170413)、溴化十六烷基三甲胺琼脂(批号170323)、肠道增菌液体培养基(批号150804)、紫红胆盐葡萄糖琼脂培养基(批号150801)、RV沙门增菌液体培养基(批号151206)、木糖赖氨酸脱氧胆酸盐琼脂培养基(批号160113)、蛋白胨(批号160801)均购自北京陆桥技术有限责任公司; 沙氏葡萄糖液体培养基SDB (批号20170429)、pH 7.0无菌氯化钠-蛋白胨缓冲液(批号20160316)购自北京奥博星生物技术有限责任公司; D/E中和肉汤(批号20170301)购自青岛海博生物技术有限公司。以上市售脱水培养基均按标签说明配制并高压灭菌后备用。
1.4 样品
抽样来自天津市A、B、C、D、E、F、G、H、I、J、K 11家医院2017年度的医院制剂, 共计41批次样品。
2 试验方法
2.1 菌液制备
取金黄色葡萄球菌、沙门菌、大肠埃希菌和铜绿假单胞菌的新鲜培养物接种至10 mL胰酪大豆胨液体培养基中, 32.5℃培养24小时; 取白色念珠菌的新鲜培养物, 接种至沙氏葡萄糖液体培养基中, 22.5℃培养72小时。取上述4个菌的新鲜培养物, 用pH 7.0无菌氯化钠-蛋白胨缓冲液制成不大于100 cfu·mL-1的菌悬液。
2.2 试验方法
本次抽样检查, 微生物限度均按照抽检医院提供的方法进行, 这些方法均是依据中国药典2015年版四部通则1105和1106建立的。在试验过程中, 依据药典要求在控制菌检查部分, 加入按照"2.1菌液制备"中制备的相应的阳性菌种进行试验。
3 结果
3.1 按照制剂剂型进行分类结果分析
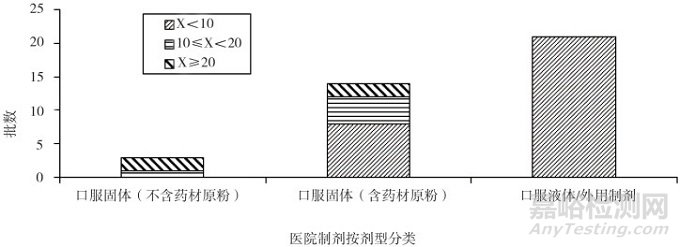
41个批次样品的生产日期均为2017年2月之后, 微生物限度标准均应符合《中国药典》2015年版四部通则1107的要求。除去3批样品因为制剂生产方(医院K)未提供2015年版《中国药典》方法适用性试验资料, 未做检查外, 具有有效检查数据的为38批。38批样品按照制剂类型及限度分为口服固体制剂不含药材原粉, 口服固体制剂含药材原粉, 口服液体制剂及外用制剂。试验结果表明:从表 1、图 1、表 2、图 2可以看出, 口服固体制剂不含药材原粉类, 3批样品均有微生物检出, 且需氧菌总数、霉菌和酵母菌总数结果偏高, 表明存在一定的微生物污染风险。口服固体含药材原粉类, 总体分布比较平均, 需氧菌总数计数93%以上都控制在200 cfu·g-1以内, 霉菌和酵母菌总数计数86%以上都控制在20 cfu·g-1以内, 因这类药品有药材原粉入药, 微生物控制难度较大, 因此有微生物生长也属于正常现象, 虽然药典规定限度较为宽泛, 但也需要严格控制。口服液体和外用制剂类, 微生物污染情况较好, 需氧菌总数计数81%以上都控制在小于10 cfu·g-1·mL-1以内, 霉菌和酵母菌总数计数均控制在小于10 cfu·g-1·mL-1以内, 外用制剂类多为化学药类, 生产工艺与中成药类相比, 微生物污染更容易控制一些。
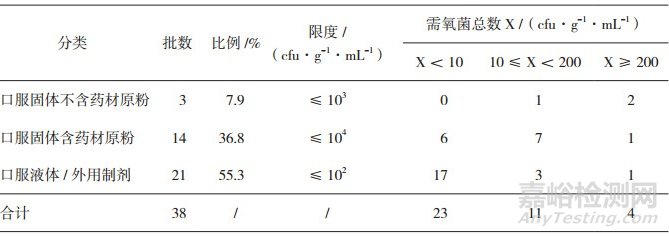
表 1 38批医院制剂需氧菌总数计数(TAMC)按剂型分类分布
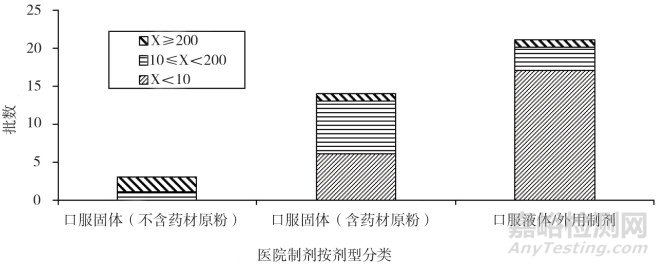
图 1 38批医院制剂需氧菌总数计数按剂型分类分布图
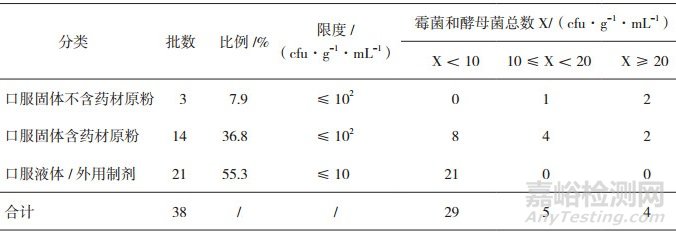
表 2 38批医院制剂霉菌和酵母菌总数计数(TAMC)按剂型分类分布
38个批次的医院制剂按照生产医院进行分类, 并分别按照需氧菌总数X < 10、10≤X < 200、X≥200 cfu·g-1·mL-1的批次数进行评价, 结果见表 3。
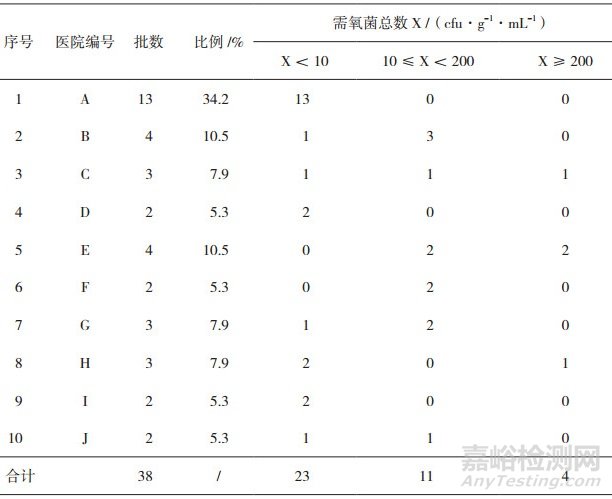
表 3 38批医院制剂按生产医院分类需氧菌总数计数(TAMC)分布表
医院A抽检的均为化学药(包括抗生素类), 占总数的34.2%, 化学药类样品, 微生物污染控制相对容易。由医院A、D、I生产的制剂, 需氧菌总数计数结果均 < 10 cfu·g-1·mL-1, 微生物污染情况较好。其余医院抽检的样品均为中成药, 占总抽样数的63.2%。医院B、C、F、G和J生产的制剂, 有部分样品有微生物生长, 检出率较高, 但菌数均偏少, 微生物污染情况较好。由医院E生产的其中3个品种为口服固体制剂不含药材原粉, 试验结果表明, 需氧菌总数计数、霉菌和酵母菌总数计数结果都有菌生长, 且补肾调冲颗粒(I)和补肾调冲颗粒(Ⅱ)的需氧菌总数计数分别为1.9×103cfu·g-1和1.6×103 cfu·g-1, 五丹胃福颗粒的霉菌和酵母菌总数计数为1.9×102 cfu·g-1, 虽没超过限度标准, 但均已接近可接受标准的上限, 表明存在一定的微生物污染, 说明医院E在制剂生产管理上存在一定的问题。医院H生产的抗白乳膏需氧菌总数检验结果为5.0×102 cfu·g-1, 超过限度标准, 结果不符合规定。
38个批次的医院制剂按照生产医院进行分类, 并分别按照霉菌和酵母菌总数X < 10, 10≤X < 20、X≥20 cfu·g-1·mL-1的批次数进行评价, 结果见表 4。
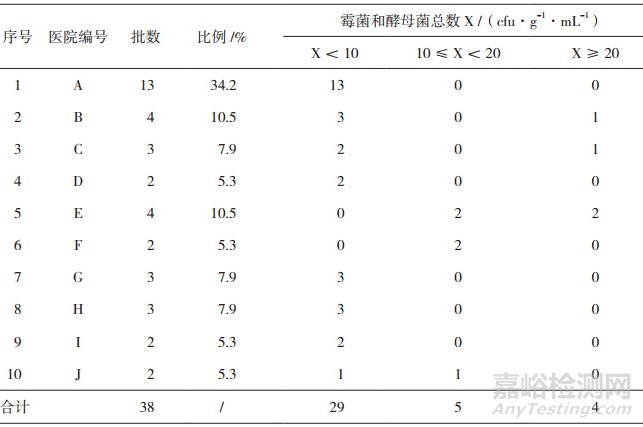
表 4 38批医院制剂按生产医院分类霉菌和酵母菌总数计数(TYMC)分布表
由医院A、D、G、H和I生产的制剂, 霉菌和酵母菌总数计数结果均 < 10 cfu·g-1·mL-1, 微生物污染情况较好。医院B、C、E、F、和J生产的制剂, 有部分样品有微生物生长, 检出率较高, 但菌数均偏少, 微生物污染情况也相对较好。
本次抽样, 从结果看, 中成药污染微生物的批数明显高于化学药(包括抗生素类), 占所有中成药总数的62.5%, 且菌数都偏多。医院H生产的抗白乳膏需氧菌总数超标, 该品种为皮肤外用制剂, 检验结果为5.0×102cfu·g-1, 超过限度标准, 结果不符合规定, 其余样品检验结果均符合规定。霉菌和酵母菌总数计数检验结果均符合规定, 控制菌检查均符合规定, 总体合格率为97.4%。
4 讨论及分析4.1 医院制剂存在的主要问题4.1.1 包装存在一定的问题
养心口服液、乳酸依沙吖啶洗剂、硫皂洗剂的瓶盖密闭性较差, 详见图 3、图 4、图 5。抗纤丸、软肝化纤丸和利肝丸的包装均为塑料袋塑封, 极容易破损, 且在包装袋外还发现了药渣的残留痕迹, 见图 6。这样的包装在运输过程中极容易因外力的影响而引起微生物的污染。

图 3 养心口服液包装瓶盖


图 5 硫皂洗剂包装
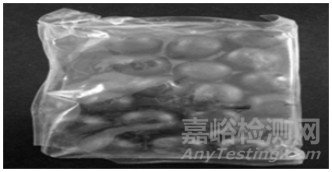
图 6 抗纤丸、软肝化纤丸和利肝丸包装
目前医院制剂原料成本逐年升高, 人工费用也不断提高, 导致医院制剂室运营成本不断升高, 有可能导致药品包装简陋。
4.1.2 检验方法不全面
控制菌阳性不生长, 缺少必要的几项控制菌检查, 以及检验方法未按照药典规定进行升级。
本次抽检所依据的检验方法均为制剂生产单位提供。根据其提供的方法, 在进行控制菌检查时, 发现部分样品的控制菌阳性对照不生长, 如硫皂洗剂的铜绿假单胞菌; 乳酸依沙吖啶洗剂(规格200 mL, 500 mL)的金黄色葡萄球菌; 氧化锌搽剂的金黄色葡萄球菌; 盐酸环丙沙星洗剂(规格100 mL, 500 mL)的金黄色葡萄球菌和铜绿假单胞菌; 后对方法重新进行修订, 修订后的方法均扩大了培养基体积并添加了聚山梨酯80和卵磷脂等中和剂, 例如, 乳酸依沙吖啶洗剂, 原金黄色葡萄球菌的检查方法为1:10供试液10 mL置300 mL TSB中进行检查, 修订后的方法改为1:10供试液10 mL置800 mL含5%聚山梨酯80的TSB中进行检查。
乳酸依沙吖啶洗剂(适应证:消毒杀菌, 用于治疗感染、化脓性皮肤病, 也可用于口腔含漱); 碘甘油涂剂(适应证:用于口腔黏膜感染, 牙龈炎, 牙周炎, 冠周炎及牙周洁治后龈袋消炎); 复方盐酸麻黄碱滴鼻液、小儿盐酸麻黄碱滴鼻液、复方薄荷脑滴鼻液3个品种均为鼻用制剂。根据《中国药典》2015年版四部通则1106表 1非无菌化学药品制剂、生物制品制剂、不含药材原粉的中药制剂的微生物限度标准口腔黏膜给药制剂、齿龈给药制剂和鼻用制剂项下规定, 控制菌检查为不得检出大肠埃希菌、金黄色葡萄球菌和铜绿假单胞菌。而制剂生产方控制菌没有进行大肠埃希菌检查, 经过试验, 增订了大肠埃希菌的方法适用性资料, 并对样品进行检查。
医院K抽样的生产日期均在2017年2月份之后的3批样品, 因制剂生产方无法提供2015年版药典的方法学适用性资料, 故未做微生物限度检查。
这些问题出现在几个医院所提供的方法中, 反映出医院制剂室在微生物方法适用性建立及药典理解方面存在一定的差距和问题。目前, 制剂室主要技术人才均是药学专业背景, 微生物专业背景的人员很紧缺, 部分从事制剂配制生产的人员还缺乏专业的学习和培训。且许多制剂室对制剂微生物控制方面是囿于环境控制不力, 因为微生物检验需具备特定的检验区域。本次抽样检查过程中, 在与医院制剂室的相关工作人员进行沟通后, 发现很多从事微生物检验工作的人员均不是专职, 还身兼其他理化方面的检验, 对微生物检验方面仅是停留在基本检验, 并不能深入研究。因此, 微生物方面的检测能力偏弱, 因而出现各种问题。
4.1.3 微生物检出率较高
虽然此次检验结果, 除1批样品需氧菌总数超过标准规定, 其余的样品检验结果均符合规定。补肾调冲颗粒(Ⅰ)、补肾调冲颗粒(Ⅱ)、五丹胃福颗粒、清热解毒片等, 虽然结果低于限度要求, 但是也已经比较接近限度值, 存在一定的安全风险。中成药中普遍菌数偏多, 导致控制菌检查在分离选择性平板上背景菌很多, 需要将疑似的菌株均挑选出来, 进行下一步的鉴定工作, 虽然在后续的鉴定过程中, 疑似菌均未被鉴定为目标菌株, 但也不排除存在一定的用药安全风险。抗白乳膏为外用皮肤制剂, 检验结果为5.0×102 cfu·g-1, 不符合药典规定。
由于中药材和中药饮片的来源主要是植物的根茎叶, 原料微生物污染情况不好控制, 如果生产工艺控制不好, 容易造成微生物的污染, 导致微生物限度检查不合格。中成药制剂在微生物控制方面, 目前均采用钴-60辐射灭菌的方法进行处理, 经过钴-60照射后, 大部分微生物被杀灭, 少部分微生物处于缺损状态, 由于中药制剂富含微生物生长所需的多种营养成分, 经过一段时间的修复, 部分微生物又能重新繁殖。
造成微生物检出率高的因素是多方面的, 有可能是包装材料的污染, 也有可能是纯化水的影响, 还有可能是生产工艺差, 操作人员在生产过程中没有严格执行操作规程而引起的。这些充分说明了医院制剂室在制备、工艺、生产及质量控制各个环节存在一定的问题, 因此, 医院制剂室要定期对药品原材料、纯化水、日常生产环境进行微生物抽查。提高全体员工防控微生物污染的风险理念, 各级领导应引起重视。
4.1.4 微生物检验结果精度不够
在微生物检验结果精度方面, 如果样品的稀释度较大, 导致样品的结果精度较低, 可以采用增加同一稀释度的平皿数, 合并计数, 以提高样品检验结果的精度。例如, 如果样品制成1:10供试液, 取1:10供试液1 mL注皿, 结果精度为 < 10 cfu·g-1(mL), 如果取1:10供试液5 mL等量分注5皿, 每皿1 mL, 合并5皿的计数结果, 结果精度为 < 2 cfu·g-1(mL)。本次抽检口服液等品种, 微生物限度标准较严格, 尤其是霉菌和酵母菌总数的标准, 为每mL中不得过10 cfu。但医院制剂所提供的方法其最低检出限为10 cfu·mL-1, 接近标准限度。虽然在方法上并没有问题, 药典也没有硬性要求, 但我们可以把工作做得更仔细、更完善, 提高计数结果的精度, 减少漏检的风险, 使检验结果更能反映真实的污染情况。
4.2 微生物限度计数方法也可能存在问题
从本次抽检的结果来看, 控制菌有6批样品阳性未生长, 不排除需氧菌总数计数及霉菌和酵母菌总数计数方面菌数回收率达不到药典规定的50%~200%。此次抽样未作复核试验, 因此所检查的结果中 < 10 cfu·g-1(mL)的样品, 不排除微生物的污染, 也有可能是该检验方法本身存在问题, 即在该检验条件下, 药品的抑菌性并没有完全消除, 不能有效地检验出所污染的微生物, 所检药品存在一定的微生物污染风险, 患者用药存在一定的安全隐患。
通过对本次医院制剂进行抽查, 集中表现出如上述所述的各种问题, 也是很多制药企业质检科室存在的问题。检验人员对药典的正确理解及执行操作是保证药品微生物限度正确检验的关键, 微生物污染理念的良好建立是制药环境得到控制最大的保障。
4.3 改进措施
针对此次抽样微生物限度检查的结果, 建议从以下两个方面进行改进。
4.3.1 加强从业人员培训
药品的微生物检验是一门理论与实践并重的学科, 加强从业人员微生物检查理论知识方面的培训, 更要重视实践技能的训练, 以期提高专业人员的职业技能, 更好地保证药品制剂的质量。
4.3.2 制剂合并生产并尝试走委托配制之路
制剂中心采取多家医院集中配制、集中管理、充分利用资源加强研究力量, 将现有的优势医院制剂开发为新药, 利用医院制剂室的条件积极开展临床药学服务等。目前, 天津市的医院制剂在合并生产、委托配制方面正在经历长足的发展, 本次抽检的医院I和医院J的制剂品种, 均是委托另一个中医院制剂室配制, 这些制剂不论从外包装还是微生物污染情况来看, 均是非常好的。这是一种资源整合的趋势, 也是很好的势头, 既保证了质量, 也提高了效率。
保证药品微生物污染水平控制在规定的限度内, 不能依赖于最终产品的微生物限度检查, 而是取决于生产过程中采用合格的灭菌工艺、严格执行《医疗机构制剂配制质量管理规范》, 并进行标准化的操作流程。这些体系的建立和保证任重而道远, 唯有此, 才能保证医院制剂微生物污染情况得到控制, 制剂质量得到更好的提高和保证。
张文燕 , 郭福庆,天津市药品检验研究院

来源:AnyTesting


