您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2022-01-13 21:53
开展强制降解试验是获得药物可能的降解途径和降解产物信息的重要途径,有助于更全面了解产品的杂质谱。对于分析方法的开发和质量标准的建立具有重要意义,是杂质检测方法建立、优化和验证的重要研究手段。可为药品的处方、工艺、包装、贮藏条件的确定提供有益支持。
1 关于强制降解的程度
一般5%~20% 的降解较为合适,需避免二次降解。对于难以降解的、非常稳定的化合物,应提供合理解释和判断依据。《化学药物杂质研究技术指导原则》中也指出,破坏试验的程度暂无统一要求,一般以强力破坏后主成分的含量仍占绝大部分为宜。此时已产生了一定量的降解产物,与样品长期放置的降解情况相似,考察此情况下的分离度更具有实际意义。要达到这种破坏程度,需要在研究过程中进行摸索,先通过文献调研、理论分析或预试验了解样品对光、热、湿、酸、碱、氧化条件的基本稳定情况,然后优化确定破坏性试验条件(如光照强度、酸碱浓度、破坏的时间、温度等),以得到能充分反映降解产物与主成分分离的结果和图谱。
关于破环试验的降解量。通常认为降解量应在5-20%之间。对于含量限度为标示量的90%-110%的小分子药物,即使在文献中有更广泛的推荐范围作为参考(例如,10-30%),通常允许10%的降解量。对于非常稳定性的药物,如何破坏都不降解,还遇到这样的发补:请继续加强对制剂降解途径和降解杂质的研究。怎么办。
2 关于强制降解的时间
(1)加强对制剂降解途径的研究
药物被破坏达到一定的时间后依然没有降解,无需再研究。,多少时间,10天,见下表(来源WHO)。
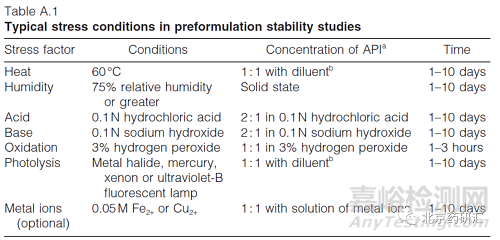
(2)加强对降解杂质的研究
从数个项目发补内容可知,破坏试验产生的降解杂质是否研究,CDE也不统一。即使不统一,研究阶段还是要研究(没有研究透彻也不影响申报,申报以后再继续研究,发补时提交资料)。加强对降解杂质的研究,言外之意是鉴定杂质结构。有一个简化的做法,参考FDA QBD模板(见下表),杂质F是未知杂质(仅采用RRT定位),限度超过鉴定限,也不鉴定结构,为什么。通过HPLC保留时间、UV(DAD),以及MS(分子量),确定杂质F是自制品与RLD的共有杂质,,没有安全性风险。
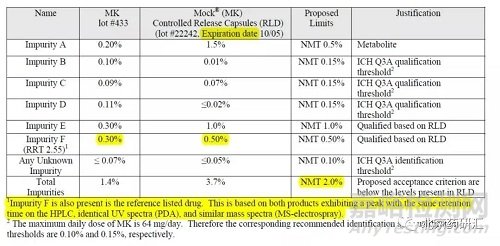
超过鉴定限的未知杂质只需用HPLC保留时间、UV(DAD),以及分子量(一级和二级质谱图),证明与RLD为共有杂质,则无需鉴定结构。
(3)氧化破坏的破坏方式
双氧水氧化(常见)、金属离子催化(在Fe3+, Ni2+, Cu2+的溶剂,通氧气使溶液中的氧处于饱和状态)、饱和氧环境(通氧气使溶液中的氧处于饱和状态)
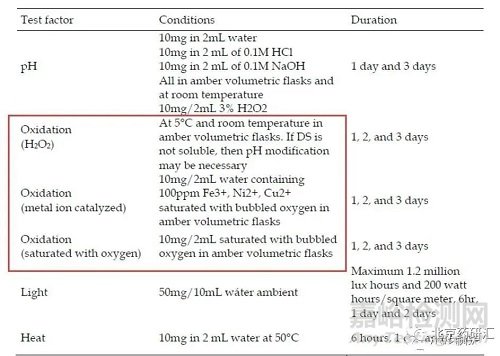
(4)酸/碱水解
水解反应是物质与水发生的导致物质发生分解的反应,即物质与水中的氢离子或者氢氧根离子发生反应。大多数有机化合物的水解,仅用水是很难顺利进行的,一般需在碱性或酸性条件下进行,这是由于水解反应的发生需克服水解基团的水解活化能(activation energy,Ea),即需要考虑水解基团所处的电子和空间位阻效应。如药物分子中含有的羧酸酯、羧酸酰胺及磷酸酰胺等基团通常水解活化能较低(Ea 通常< 20 kcal·mol-1 ),是比较容易水解的位点; 如醚及磺酰胺基团,一般水解活化能较高(Ea 通常> 30 kcal·mol-1 ),通常不易发生水解。
酸/碱水解考察条件的设置主要考虑因素包括:酸/碱溶液的浓度(或pH 值)、考察的温度与时间,具体考察条件需根据药品特点,特别是分析药物结构中含有的水解基团及其所处的电子和空间位阻环境。如对于含有羧酸酯的药物,其可能对碱水解十分敏感,就可使用较低浓度的氢氧化钠溶液,在室温条件下进行考察即可。而同样对于含有羧酸酯的药物,如果所处空间环境位阻较大,如叔丁基酯,可能水解条件需适当加强。
另外,一些在水溶液中溶解度不好的亲脂性药物,需注意添加适当的有机溶剂进行增溶,不应一味地增大破坏强度,造成次级降解。常用水解考察条件包括: 0.1~1 mol·L-1 的盐酸或氢氧化钠溶液,在室温或加热条件下进行考察,如60 ℃ /2 d。
(5) 光照降解
光照强制降解试验的条件设置在ICH Q1B[10]中有较明确规定,可分别在样品均质化或溶液状态下进行考察,一般要求总照度不低于1. 2 × 106 Lux·h(冷白光灯)或近紫外能量不低于200 w·h·m-2(紫外灯),如254 或365 nm 光源照射24 h。
需注意将光照发热对受试样品的影响降到最低,还应考虑样品的物理性质,并应采取措施如冷藏和/或置密闭容器中,以确保物理状态变化(如升华、蒸发、熔化)所造成的影响最小。
(6) 高温降解
对于热降解,一般遵循阿伦尼乌斯(Arrhenius)方程,即随温度升高降解速率加快。高温降解试验即运用这一原理,通过设置较高的考察温度在较短的时间内获得药品的降解信息。具体考察温度和时间需根据药品特点,在前期预试验的基础上灵活确定,常见如80 ℃ /10 d,130 ℃ /8 h。也常结合湿度进行设置,如对于原料药或固体制剂通常采用相对湿度75% 或更高(如80 ℃ /92. 5% RH等),而对于液体或半固体制剂可能需考虑采用干热条件(低湿度),如相对湿度25%或更低。受试样品可分别在固体和溶液状态下进行考察,需注意涵盖生产过程中最差条件的考察,如含主药的料液在喷雾干燥过程中温度升高,又如半固体制剂水相或油相溶解主药过程中可能需升高温度。对于破坏程度过高或过低的情况,可能还需进一步结合更接近于实际高温情况的影响因素试验(如60 ℃ /75%RH/1 个月)的考察结果,来综合判断是否药物本身对热特别稳定或特别敏感。
(7)物料平衡
下表是两种计算方法有不同的角度,但不仅限于两种,可自行选择适合的计算
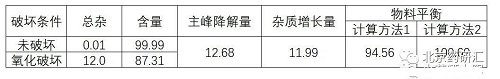
计算方法1:杂质增长量÷主峰降解量×100
计算方法2:100-主峰降解量+杂质增长量
如果存在物料不守恒(含量下降5%,总杂小于1%),通常有两个假设:
假设1:溶液提取不充分。
思路:通过不同溶剂提取(酸、碱、甲醇、乙腈、DMSO等)、超声的时间和温度提取。
假设2:降解物未检出。
思路:降解物高水溶液-无保留,采用亲水色谱体系;高脂溶性-未洗脱,采用高比例有机相梯度洗脱;无紫外吸收,更换检测器如ELSD、CAD等。
备注:各已知杂质与主峰的分离度满足要求的情况下,含量测定方法验证无需考察破坏试验

来源:北京药研汇


