您当前的位置:检测资讯 > 科研开发
嘉峪检测网 2021-02-26 15:59
近年来,基因毒问题是药物开发、乃至上市后药品质量评价的重要内容之一,甚至成为了行业内的“闪点”、“燃点”!然而,大部分同行在聊及基因毒问题时,大都仅停留在警示结构、1.5ug的概念,很少从药理毒理的角度来思考基因毒的真正内涵。故整理此文,望共同进步。
01、什么是遗传毒?
遗传毒性可分为DNA损伤、基因突变、染色体结构改变和染色体数目改变四类。广义的遗传毒性,是指由遗传毒物引起生物细胞基因组分子结构特异改变而使遗传信息发生变化的有害效应;因此,通过DNA损伤产生突变以及基因组复制过程误差率增高皆为遗传毒性的表现。而狭义的遗传毒性是指遗传毒物对DNA(或染色体)的损伤作用。
02、遗传毒理学历史
1927年,遗传毒理学起源于H.J.Muller的开创性工作,他的研究发现放射不仅能够增高突变的频率,而且诱发的突变型或表型与没有放射时所观察的完全一致。所以,诱发的突变反应必须根据基础突变来评价。
1938年,Sax在Muller研究的基础上,又研究发现X射线能够诱发紫露草花粉颗粒中染色体结构畸变。在完全缺乏有关DNA结构和染色体组成的情况下,Sax和他的同事发现染色体内或染色体间的交换需要在核内至少有两处大的损伤。另外发现,接触X射线总剂量不变,但延长接触时间,或在几小时内分为两部分接触,产生染色体畸变的量降低。
1946年,Auerbach和他的同事们报道了氮芥能诱发果蝇突变,这些突变与X线诱发的突变表型是相同的,由此人们开始考虑化学物质对遗传的作用。
到了20世纪70年代,有两个事件使得能用诱变性资料来评价危险度。Miller和他的同事们研究发现化学致癌物能在体外和体内与DNA、RNA及蛋白质作用形成稳定的共价衍生物。
同一时期,第二次发展改变了遗传毒理学领域,那就是Ames(1975)和他的同事们所建立的用伤寒沙门菌进行一种简单、廉价的突变试验;该实验可检测化学物所致的组氨酸基因座的回复突变,并能结合使用一种外援性S9代谢系统。
Ames试验于今天已被广泛使用,而进一步同为20世纪70年代的体内微核试验,经过几十年的发展和应用,目前已出现使用图像分析、流式细胞仪和激光扫描细胞仪等多种自动化系统检测微核的方法。
再之后,1994年Gatehouse等在Mutation Research上发表了国际遗传毒性试验标准化研讨会上制定的开展Ames试验的一系列建议;且OECD于1997年制定了Ames试验的指导原则,又进一步促进了该方法的标准化和应用。
而今,ICH、OECD等机构已制定并颁布多个指导原则,用于遗传毒理学的研究,并在不断的持续更新……
03、我国国内发布的指导原则
我国于2007年10月23日发布了《药物遗传毒性研究技术指导原则》。在该指导原则起草过程和审评过程中一直强调:指导原则只是原则性指导,且仅能反映当时的认识!研究者不能机械地照搬指导原则,而是应充分发挥主观能动性,积极跟踪学科进展,推动研究工作。
2018年3月,为指导和规范药物遗传毒性研究,国家食品药品监督管理总局组织修订了《药物遗传毒性研究技术指导原则》,且同时宣布原国家食品药品监督管理局2007年发布的《药物遗传毒性研究技术指导原则》废止。
标准试验组合
指导原则指出:根据试验检测的遗传终点,可将检测方法分为三大类,即基因突变、染色体畸变、DNA损伤;根据试验系统,可分为体内试验和体外试验。标准试验组合应反映不同遗传终点,包括体内和体外试验,应包含以下内容:1)应包含细菌回复突变试验(又称Ames试验),该试验已证明能检出相关的遗传学改变和大部分啮齿类动物和人类的遗传毒性致癌剂;2)应包含哺乳动物细胞体外和/或体内试验。
表1:国内指导原则标准试验组合

04、ICH颁布的指导原则
1995年和1997年ICH分别颁布遗传毒性指导原则S2A(药物遗传毒性试验的特殊性指导原则)、S2B(遗传毒性:药物遗传毒性试验标准组合);ICH遗传毒性指导委员会于2006年6月启动了遗传毒性指导原则的修订工作,并多次在美国、欧盟和日本等地召开会议商讨修订工作,最终于2011年11月正式修订完成,完成ICH第四进程,推荐给ICH管理三方(欧盟、美国、日本)采纳使用。
标准试验组合
指导原则指出:选择一具有更多的历史应用经验,部分原因是由于该组合被S2A和S2B指导原则推荐。然而,认为选择一和选择二同等适合的原因如下:当体外哺乳动物细胞试验结果为阳性时,两个实施良好的体内试验(采用合适的组织和显示有充分的暴露)的明确阴性结果,被认为足以证明缺乏体内遗传毒性潜在性,因此进行两种体内试验的试验策略,与对体外试验阳性结果进行追加试验是相同策略。
表2:ICH S2(R1)指导原则标准试验组合

05、历史上发生的药品基因毒代表性事件
2007年6月,罗氏制药在欧洲市场推出的HIV蛋白酶抑制甲磺酸奈非那韦,因为在其产品中发现一种经典的基因毒性杂质~甲基磺酸乙酯超标,EMA暂停了它在欧洲的所有市场销售。甲磺酸乙酯的引入是由于在生产设备清洗时,乙醇未被完全烘干而残留下来,与甲磺酸奈非那非原料药中的甲基磺酸反应形成甲基磺酸乙酯。
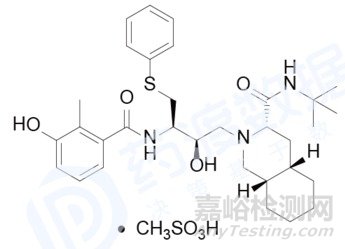
甲磺酸奈非那韦化学结构
罗氏在被要求彻底解决污染问题后,还需要补充毒性研究数据,以更好的评估甲磺酸奈非那非的基因毒杂质对患者的风险。直到罗氏公司完全解决了这些问题后,EMA才恢复了甲磺酸奈非那非在欧洲市场的相关授权。在这次甲磺酸奈非那非事件发生之后,世界各国的药品管理机构均针对基因毒性杂质提出了更加明确的规定和要求,因此,国内外医药企业在新药的研发过程中也越来越重视基因毒杂质的控制和检测。
06、小分子药物中常见的基因毒(杂质)问题
那么,如何认识和控制基因毒杂质呢?
基因毒杂质的结构多种多样,对于绝大多数的杂质而言,往往没有充分的毒性或致癌研究数据,因而难以对其进行归类。在缺乏安全性数据支持的情况下,大多数法规和指导原则采用“警示结构”作为区分普通杂质和基因毒性杂质的标志。对于含有警示结构的杂质,应当进行(Q)SAR预测和体内外遗传毒性和致癌性研究,或者将杂质水平控制在毒理学关注阈(TTC)之下。
关于基因杂质警示结果的具体详细信息可参考欧盟发布的警示结构《Development of structural alerts for the in vivo micro nucleus assay inrodents》,或进入The Carcinogenic Potency Database(CPDB),里面有上千种致癌物质的列表、结构式、CAS号,作用部位,TTC值等一系列信息。
传说中超级致癌物的虚拟结构(CNKI)
ICH最早给出了的化学原料药杂质研究指导原则Q3A(R2)、制剂杂质研究指导原则Q3B(R2),在这些指导原则中首次提到“对于能够产生强的药理活性或毒性的潜在杂质,即使其含量低于0.1%,也必须进行进一步的结构鉴定”。在之后修订的版本中,进一步明确“要关注原料药中的潜在遗传毒性杂质”,以及“对于毒性非常强的基因毒性杂质,需要根据实际情况来制定更低的检测限度”,但也并未针对遗传毒性杂质及其研究和控制问题提出明确的阐述,同时,对其研究原则、限度要求及控制策略均没有具体的要求。
EMEA人用药品委员会(CHMP)推出《遗传毒性杂质限度指导原则》,引入了可接受风险的摄入量,即毒理学关注阈值(TTC)这个概念。设置了限度值TTC(1.5 μg/day),即相当于每人每天摄入1.5 μg的基因毒性杂质,被认为对于大多数药品来说是可以接受的风险(一生中致癌的风险小于十万分之一)。根据此阈值,按照每日预期的摄入量去计算出该活性药物中能够接受的杂质水平。值得注意的是,TTC只是一个概率的方法,具有一定的风险。如果存在一个基因毒性杂质,对其毒性未知,假设其每日的摄入量在TTC范围内,那它致癌的概率将会在10-5以下,因此限度值(TTC)不等于完全没有风险。PS:针对不同类型的药物,也有不同的阈值及算法。
表3:药品中的基因毒杂质分类
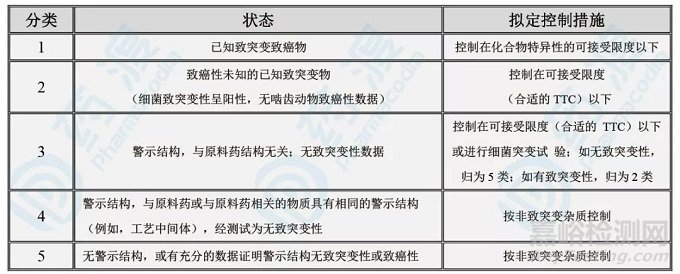
07、M7指导原则~非常重要
M7于2014年6月由 ICH 指导委员会在ICH进程的第4阶段中予以通过;在ICH 进程的第4阶段,最后的草案被推荐给欧盟、日本和美国的监管机构采纳。
M7旨在提供一个可用于致突变杂质的鉴别、分类、界定和控制的可行性框架,以限制潜在的致癌风险。同时,意在补充 ICH Q3A(R2)、Q3B(R2)和M3(R2)支持药物进行临床试验和上市的非临床安全性研究。PS:M7不适用于ICH S9范围定义的用于晚期癌症适应症的原料药和制剂。
M7再次给出了TTC、TD50、1.5μg/天等概念和数据,同时重点强调了上市后药品变更后的基因毒杂质问题;见下表。
表4:上市后药品变更所要面对的基因毒杂质问题
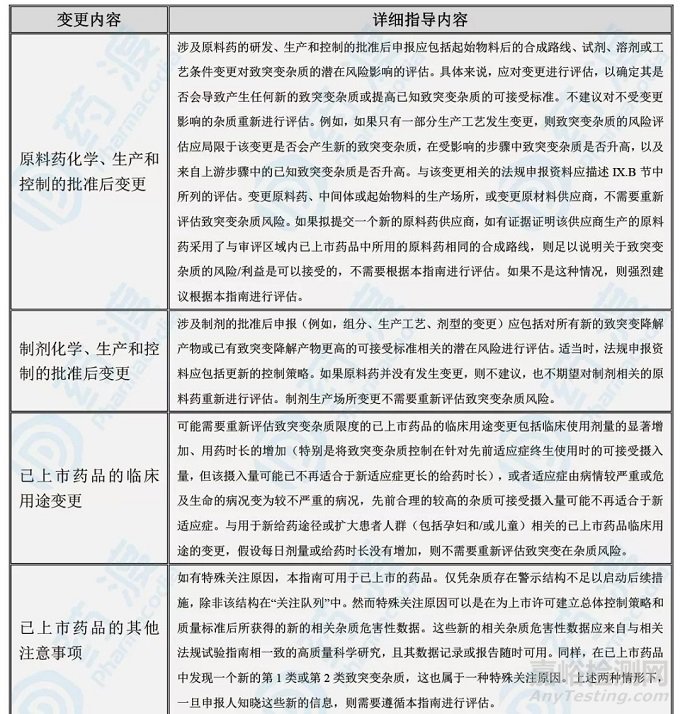
08、结语
基因毒问题,最为根本的还是安全性问题,而一个物质是否安全、是否存在基因毒,要靠试验和数据来证明。至今,我们已经知晓了很多基因毒物质、也知道了怎么判断警示结构,但依然出现了NDMA事件!且不论事出原因如何,结果都是药品的质量出问题了!不过,当质量出现问题之时,作为一个药品从业人员,反思更多的应该是问题出在了哪里?到底我们应该如何做出“安全、有效、质量可控”的药物!
综上,即为笔者当下理解的遗传基因毒内容。
参考:
1. ICH 三方协调指导原则 .
2. 人用药物遗传毒性试验和结果分析指导原则S2(R1).
3. ICH.S2(R1):Guidance on genotoxicity testing and data interpretation for pharmaceuticals intended for human use. 2011
4. ICH.M3 (R2): Non-clinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals. 2009
5. FDA. Guidance for industry and review staff: Recommended approaches to integration of genetic toxicology study results. 2006
6. OECD. Guideline for testing of chemicals No.471:Bacterial reverse mutation test.1997
7. OECD. Guideline for testing of chemicals No.473:InVitro mammalian chromosomal aberration test. 2016
8. OECD. Guideline for testing of chemicals No. 488:Transgenicrodent somatic and germ cell gene mutation assays. 2013
9. OECD. Guideline for testing of chemicals No. 489:Invivo mammalian alkaline comet assay. 2016
10. OECD. Guideline for testing of chemicals No. 490:Invitro mammalian cell gene mutation tests using the thymidine kinase gene. 2016
11. M7 评估和控制药物中 DNA 反应性(致突变)杂质以限制潜在的致癌风险.
12. M7(R1):评估和控制药物中DNA反应性(致突变)杂质以限制潜在致癌风险.
13. 遗传毒性杂质的警示结构.CNKI
14. http://samr.cfda.gov.cn/WS01/CL0087/226424.html
15. 书籍:《毒理学》-毒物的基础科学

来源:药渡


