非临床安全性研究很重要的一个目的是确定药物的毒性靶器官、毒性表现,并发现可用于计算首次人体试验的剂量,支持药物临床开发。NOAEL(No Observed Adverse Effect Level,未见明显毒性反应剂量)在健康成年志愿者首次临床试验药物最大推荐起始剂量估算过程中发挥主导作用。除了NOAEL,MABEL(minimal anticipated biological effect level)、HNSTD(highest non-severely toxic dose)、STD10(severely toxic dose in 10% of the animals)也被用来作为某些药物首次人体试验起始剂量的计算。无论何种计算路径,对于非临床研究中的异常发现是adverse、non-adverse的判定都是核心,而这一鉴别又颇具挑战,有时偏主观。本文结合FDA、Amgen、Gilead、Celldex Therapeutics等团队最新观点,围绕毒性反应、NOAEL判定原则、挑战、案例进行分享。
首先看下对于adverse effect的定义,来自两个版本。
European Society of Toxicologic Pathology: An adverse effect is a test item-related change in the morphology, physiology, growth, development, reproduction or life span of the animal model that likely results in an impairment of functional capacity to maintain homeostasis and/or an impairment of the capacity to respond to an additional challenge.
The Illustrated Dictionary of Toxicologic Pathology and Safety Science: adverse effects (in toxicology studies) are any changes related to treatment with a test substance that are considered as potentially harmful to the well-being of the test species, resulting in dysfunction, or negatively impacting the ability of an animal to thrive or develop normally.
两个版本来源的定义描述不同,但底层逻辑类似,首先是需要发现一些changes,或许是与平行对照比,或者与历史背景数据比,或者通过剂量依赖性关系判定。其次,需要对发现的changes进行定性,是否影响了动物的某些功能、生长、发育等等。
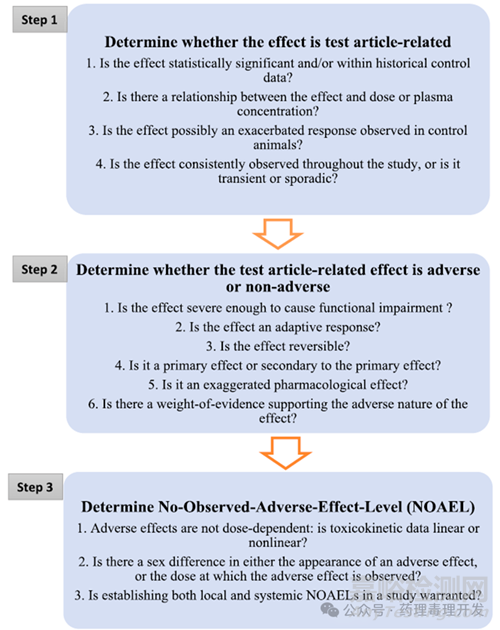
关于NOAEL的确定,通常采用“三步法”,如下图所示。
第一步:确定是否是供试品相关的作用
关于这点,可以从4个问题驱动,辅助判断。
1、是否在历史对照数据范围内和/或具有统计学意义?
对于定量数据,首先应进行统计学分析,看下高剂量与对照组、低剂量之间的统计学差异。尤其是啮齿类动物,每组动物数量多,且遗传同质性好,更适用于均值比较和统计分析。对于非啮齿类动物,由于每组动物数量少,个体动物发现有时比均值更有意义。建议从总体证据考量发现的毒性,而不是单一指标的变化,比如肝脏生化指标的变化是否伴随大体解剖、组织病理学检查的异常。
当然,并不是所有毒理学终点都可进行统计运算,如组织病理学检查中的发生率和严重程度通常不进行统计分析。类似这种情况,需要结合平行设置的对照组表现和历史对照数据进行结果判定。这里就比较考验每个机构历史对照数据的质量了。
2、是否有剂量/暴露量-反应关系?
虽然不绝对,但绝大多数毒性发现,如果无剂量或暴露量相关性,与供试品相关的可能性通常较低。从药代动力学角度,药物暴露量(AUC或Cmax)随着给药剂量升高而升高,但涉及吸收或消除饱和,剂量和暴露量的升高不一定成比例。一般毒理研究中会伴随毒代动力学研究,可同时结合剂量/暴露量数据,分析与毒性发生率、严重程度的相关性。特别提示两点,有些毒性反应如功能组合试验(functional observational batteries)、心电图、血压对血药浓度更敏感,建议结合Cmax分析。另外,有些毒性如果仅在高剂量出现,低、中剂量未见,且暴露量是线性增加的,也提示该毒性反应为供试品相关。
3、给药组的反应是否比对照组观察到的更严重?
非临床研究用动物通常都会有背景病变,因这种病变的发生类型、发生率和严重程度不同,平行设置的对照组动物并不一定能客观反映背景病变情况。举个例子,一项6个月SD大鼠重复给药毒性试验中,雌性动物观察到频率和严重程度均呈剂量相关性的肾上腺皮质囊性变性,雄性动物呈现类似发生率的多灶性肾上腺皮质空泡化,平行设置的对照组动物未见类似病变。肾上腺异常是大鼠年龄相关性自发性病变,且SD大鼠发生率更高。最后结论是供试品有可能会加重大鼠肾上腺背景病变,但考虑到这一病变是大鼠自发性年龄相关改变,不视为毒性反应(not considered adverse)。当然,这种结论的得出需要结合研究机构的背景数据和大量文献数据进行讨论。
4、这一反应是在整个研究过程中一直存在,还是一过性的或偶发的?
这个问题适用于研究过程中反复观测的毒性终点,比如临床观察、体重、摄食量、临床病理等。如果某些临床症状仅在个别动物出现,并不是在整个研究中出现,仅是偶发的,通常不认为是供试品暴露引起的。不过,这个问题也需要辨证看,不能将所有一过性、偶发性的毒性发现,均视为非给药相关。尤其是临床病理数据,比如ALT和AST一过性升高,意味着可能的肝细胞损伤,随着给药继续,有可能肝细胞由变性进展到坏死,出现代偿性肝细胞增殖或纤维化,ALT和AST反而回到基线,不再升高。这种情况,可以结合病理或其它肝脏标记物一起判断,不可轻易排除。
第二步:确定供试品相关作用是否是毒性反应
关于这点,可以从6个问题驱动,辅助判断。
1、是否足够严重到引起功能损伤?
通常体重、摄食量和临床观察可以帮助从整体判断动物的健康状况。如果这些参数的异常关联到动物功能的损伤,则判定为毒性反应。对于什么程度的变化认定为异常,OECD发布过指导原则《Document on the Recognition, Assessment, and Use of Clinical Signs as Humane Endpoints for Experimental Animals Use in Safety Evaluation》。以体重为例,对于非减肥药物,体重降低20%以上通常认为是adverse。当然,还是那句话,不要孤立的通过一个指标轻易下结论,结合对照数据、统计学、量效关系变化、指标变化的连续性等综合分析。
通过临床病理学数据判定毒性反应是有一定挑战的。钠、钾、钙、葡萄糖、血红蛋白和血小板,视变化程度大小判定是否是毒性反应。单纯的ALT、AST、LDH变化通常不视为毒性反应,可以作为组织损伤的biomarker,需要结合其它临床病理参数及组织病理学数据共同确认。像总蛋白、白蛋白这类体现肝功能的参数,低于一定数值可判定为毒性反应。
组织病理学检查结果根据病灶病变严重程度进行分级,通常“minimal”不记为毒性反应,“marked”或“severe”则视为毒性反应。也有些特例,比如神经元坏死的“minimal”分级则视为毒性反应。
2、是否是适应性反应?
为应对有毒物质的侵犯,机体为维持稳态或产生防御,通常会出现代偿性变化,称之为适应性反应。这种适应性反应通常是快速的、可逆的,不会对机体造成损伤。比如苯巴比妥、美沙酮、纤维蛋白或肝药酶诱导剂,给药后会出现肝脏重量增加、肝变大、镜检可见肝细胞肥大,伴随多次给药后的药物暴露量下降。这类变化如果未引起其它degenerative changes,通常不视为毒性反应。
3、反应是否可逆?
并不是说部分或完全可逆的反应就不视为毒性反应,而是需要结合所有毒理学发现和证据权重分析进行综合研判。可逆性可作为整体性的一部分,辅助判断毒性发现的严重程度。
4、是供试品引发的原发作用还是继发作用?
首先,无论是药物引起的原发作用,还是由原发作用进一步引发的继发作用,都会用于评估是否是毒性反应。不过,有些继发作用可以通过人道关怀、兽医护理等得以改善。比如药物可引起犬恶心,出现流涎、呕吐等症状,继而导致摄食量降低、体重下降,通过延长食物供给时间,摄食和体重可恢复。这类情况,前期动物出现的摄食量和体重异常则不视为毒性反应。
5、是否是药理作用的放大?
药理作用的放大就一定是non-adverse吗?答案当然是否定的。还是要视药理作用放大的程度及所引发作用的严重程度而定。比如免疫激动剂可过度激活免疫系统,释放细胞因子,并可能出现动物摄食量、体重下降的情况,甚至导致动物不能耐受,则可判定为adverse effect。药理作用的分类太过宽泛,这也给药理作用放大后的adverse、non-adverse的判定带来很大挑战,比如作用于CNS的药物引发的动物平衡、协调失调,催眠类药物可使动物丧失基本能力。有时很难对这类毒性反应进行区分和定性,需要结合严重程度、发生率、发生的频率综合判定。
6、基于证据权重判断药物反应是否严重到影响器官或机体的功能
这点还是强调基于总体的证据对药物所致的毒性发现进行综合分析。通常基于毒理试验中所有可获得数据、文献资料,GLP机构的SD会对出现的药物反应的定性有个大致判断。当然,对于疑问之处,也可与同行、顾问、专家进行进一步讨论。药企sponsor对药物的整体纵深了解通常更为全面,有比较详细的药效学、药代动力学和毒理学资料,通常也熟悉同类产品的临床研究结果,故与sponsor的充分沟通也是非常关键的。
第三步:确定NOAEL
NOAEL的确定说简单也简单,比如中剂量观测到供试品相关的毒性反应,低剂量未观测到,则低剂量可定为NOAEL。不过,在具体决策过程中还是有3个问题需要思考。
1、对于毒性反应非剂量依赖的情况:暴露量是线性还是非线性?
线性PK的AUC和Cmax随剂量增加而成比例增加,表观分布容积、消除半衰期、清除率则剂量间表现一致。相反,非线性PK的AUC和Cmax与剂量的增加不成比例,表观分布容积、消除半衰期、清除率在不同剂量间也有差异。有时会出现中剂量血药浓度更高,出现毒性反应,高剂量反而血药浓度降低,未见毒性反应的情况,这种情况下的NOAEL应定为低剂量。
2、毒性反应是否有性别差异?
不同性别之间的药物PK和毒性敏感度不同还是挺常见的。如果有明确证据说明这点,就需要雌雄动物分别界定NOAEL剂量,取剂量更低者用于临床起始剂量的计算。当然,不出意外的话的还是有意外出现,比如一个药物临床仅用于男性,但毒理试验采用的双性别动物,雌性动物低剂量是NOAEL,雄性动物则是高剂量为NOAEL,这种情况自然可以选择雄性动物的剂量进行临床起始剂量的计算。
3、是否有必要建立局部和系统NOAEL?
比如药物低剂量可见注射位点反应,但无其它毒性发现。中剂量既有注射位点反应,也可见系统的毒性反应。这类情况的处理方案通常有两种,一种是低剂量定为系统NOAEL,但标注有注射位点反应。一种是继续探索更低剂量,无注射位点反应的局部NOAEL。关于注射位点反应,如果不是比较严重的情况,通常不作为限制性因素影响NOAEL的制定。一是比较容易耐受,二是临床可以给予抗组胺药或其它药物干预。
那有没有需要界定特定器官NOAEL的情况呢。比如药物IND后,监管机构对药物的心脏安全性风险有担忧,要求提供进一步数据说明。后续开展的毒理研究更多聚焦心脏重量、心脏毒性的biomarkers、心脏大体解剖、心脏的组织病理学检查,未额外开展系统的毒理学研究,那最终获得的NOAEL则是心脏特异性的。不过,这种情况并不多见。
案例分享
案例一
已上市非抗肿瘤药物增加新给药途径,开展28天大鼠静脉重复给药毒理研究。高剂量出现震颤、步态改变、呼吸困难等严重临床症状,镜检可见肝脏空泡化、垂体前叶变性。另外,可见剂量相关性的甲状腺肥大。进行了TK分析,但未开展甲状腺激素检测。
CRO SD:考虑到中剂量出现较minimal更为严重的甲状腺肥大,NOAEL定为低剂量。
Sponsor:不认可SD结论。提供了同一药物其它种属、更高剂量下、相同甲状腺病理结果的数据,显示未见体现甲状腺功能的甲状腺激素的异常。
CRO 病理学家:依然不为所动,坚持己见,认为只有大鼠的功能性数据才能说明问题,其它种属的不予考虑。
CRO高级毒理学家:查阅了该药物前期上市时开展的大鼠13周重复经口给药毒理研究数据,更高暴露量情况下,未见甲状腺激素的变化。
最终,CRO病理学家同意修改意见,判定中剂量出现的甲状腺病理改变为non-adverse,SD修改NOAEL剂量为中剂量。这个案例首先判定甲状腺肥大严重程度是剂量相关的,是与供试品有关的反应。至于判定为毒性反应还是非毒性反应,不同相关方的结论不一。本文第二步的第1个问题,是否足够严重到引起功能损伤,比较庆幸的是甲状腺异常是有功能性biomarkers可以支持决策的。也提醒NOAEL的确定不仅需要采纳本试验数据,还需结合前期数据、sponsor观点等综合考量。
案例二
ADC抗肿瘤产品,大鼠静脉给药3周毒理研究。骨髓、胸腺、脾脏、肺及睾丸均发现病理学变化,且呈剂量依赖性,低剂量严重程度是minimal/mild级别。除睾丸外,中、高剂量组动物的毒性发现经6周恢复期后可逆性恢复。
基于以上信息,CRO SD将NOAEL定为了低剂量。Sponsor认为低剂量用于计算起始剂量太低,咨询了其它毒理顾问意见。看到这,不晓得各位是否意识到其中的问题,供试品的临床拟用适应症是肿瘤,根据ICH S9要求,啮齿类动物STD10的1/10作为起始剂量,故应该根据是否引发动物死亡/濒死、威胁生命或不可逆毒性情况获取STD10,而不单纯是NOAEL。当然,最后SD对报告进行了调整。这个案例提示我们,不是所有试验都需要界定NOAEL。另外,CRO与Sponsor的充分沟通非常关键,不要等到最后试验都做完了,产品目标适应症还不清楚。
案例三
一个多肽类药物,拟用于非肿瘤适应症,皮下给药,犬9个月重复给药毒性试验。给药2个月时,中、高剂量组注射部位出现中重度红斑和水肿,而且注射位点反应已经影响到继续给药。鉴于这种情况,CRO和sponsor进行了密切沟通和反复讨论,提出了各种各样的解决方案,包括重新开展研究、增设更低剂量组动物,两个路径都涉及费用和时间的大量增加,而且在已经进行的试验期间增设组别非常麻烦。还有一种方案是因低剂量未见注射部位反应,说明该毒性表现是剂量相关的,可以尝试降低中、高剂量组的剂量解决。亦或者对于感染动物先停药,恢复后继续用药。还可以改变注射部位,换其它位点。另外,研究发现注射位点反应是组胺介导的,可以给予抗组胺药物处理,增加局部耐受。最后,经过多方讨论,采用的是给予抗组胺药+动物短暂给药休息+调整注射位点方案,顺利完成了9个月的给药周期。
最后给出了两个NOAEL,低剂量为局部NOAEL,高剂量为系统NOAEL。这个案例提示我们,试验出现问题不可怕,充分的头脑风暴、信息互通对于解决问题反而颇为重要。对于注射位点反应这点,正文中已有解释,不再赘述。
案例四
一个小分子药物,拟用于非肿瘤适应症,经口给药,大鼠6个月重复给药毒理试验。高剂量组在给药第30天,中剂量组动物在给药第100天,低剂量组在第130天出现seizures症状。有时发生在药后几秒,有时发生在采血、体重测量、换笼等日常操作过程中,持续约1分钟。每个组别seizures发生率和发生次数如下表所示。
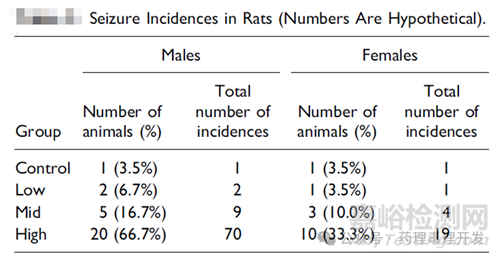
动物体重、摄食量等其它指标均正常。也未见动物发作后抑郁表现。恢复期也未见动物出现seizures。虽然未见动物死亡/濒死,考虑到高剂量组动物发作的频率较高,sponsor决定在第90天时,高剂量组动物停止给药,进行组织病理学检查,其它动物按原定方案继续用药。病理发现高剂量组脑部空泡化。结合病理结果和临床症状观察,高剂量组出现的seizures定性为毒性反应。很明显seizures发生率呈剂量相关性,是供试品相关的。但问题也来了,所有的seizures都应视为毒性反应吗?单次seizures发作算毒性反应吗?低剂量组的seizures发生率算毒性反应还是非毒性呢?关于这点,CRO公司、申报方和其它相关团队并未完全达成一致。
最后,将低剂量定为NOAEL,该组的seizures定性为可能的非毒性反应(likely non-adverse)。原因如下:1)低剂量组发生率很低,与对照组几乎相当;2)动物并未在Tmax时间点发作,更多在被操作时发作,不具备血药浓度依赖;3)发作时间歇性的,数天到数周不等;4)动物未见发作后抑郁,而是发作后快速恢复,并正常生活;5)整个治疗期间动物健康存活;6)动物未见其它指标异常。
暂不提这些原因有些是不是稍显牵强,这个案例对于中枢神经系统毒性反应相关的NOAEL界定,还是有参考价值的。
案例五
一个小分子药物,拟用于非肿瘤适应症,经口给药,大鼠3个月重复给药毒理试验。中、高剂量组动物出现肾毒性、临床症状异常(药后1h内约50%动物出现没有精神)、体重降低(平均20%)。此外,高剂量组出现动物死亡。所有剂量组动物均出现肝细胞脂质增加、肝脏重量增加、肝小叶中心肥大,严重程度呈剂量依赖性增加。提交到监管机构的资料显示,中剂量被定为NOAEL,脂质代谢异常、临床症状异常和体重降低未被视为毒性反应。当然,监管机构最终不同意这一NOAEL选择,认为应该将低剂量作为NOAEL。监管机构认为中、高剂量组转氨酶升高、肝细胞脂质增加、血浆胆固醇增加等多个临床病理指标异常均指向了脂质代谢过程的异常,应视为毒性反应。再结合肾毒性、体重降低、临床观察异常,中剂量不应作为NOAEL。
篇幅所限,更多案例有时间另起一篇再单独介绍吧。本文更多是把adverse和non-adverse判定的基本逻辑、大致过程和核心问题做下梳理,可以看到,NOAEL的确定是一项复杂、系统的技术活,需要有一套严格的决策步骤,不是完全凭主观和经验武断给出答案。更何况,NOAEL的确定涉及到临床起始剂量的选择,定的过低,需要额外爬坡,费时费力费钱。定的过高,如果不合理,监管机构不会同意,也会增加注册时长。