近年来寡核苷酸药物成为新药研发中备受关注的分子类型,被认为是继小分子化药、抗体药物之后的第三代治疗药物。寡核苷酸是指含有约20个碱基的短链核苷酸,通过以Watson-Crick碱基互补配对的原理与mRNA互补链结合,作用于转录和翻译。根据结构和作用机理不同,分为反义寡核苷酸(ASO)、小干扰RNA(siRNA)、小激活RNA(saRNA)、微小RNA (miRNA)、适配体(Aptamer)等。
由于siRNA药物疗效较好且技术取得突破,这使得它成为目前最受关注的核酸药物之一。siRNA是一类双链RNA分子(反义链、正义链),通过与体内相关酶形成RNA诱导沉默复合物(RNA-induced silencing complex, RISC)来降解目标mRNA,以此来下调或抑制目标基因的表达。2024年伊始,siRNA药物领域的合作捷报频传:1月3日,勃林格殷格翰宣布与苏州瑞博及其瑞典子公司Ribocure达成合作协议,共同开发治疗非酒精性或代谢功能障碍相关脂肪性肝炎的小核酸创新疗法。1月7日,舶望制药宣布与诺华就RNAi疗法达成两项独家许可合作协议。
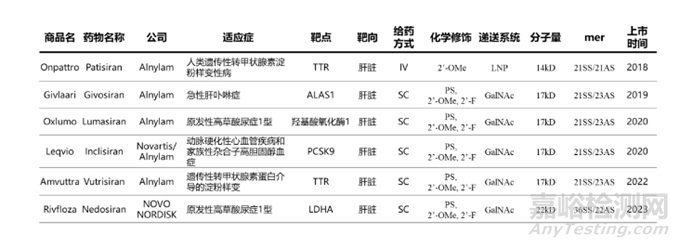
截至目前,FDA共批准6款siRNA上市药物。值得一提的是,其中有4款来自Alnylam,1款来自Alnylam与诺华共同开发,1款来自诺和诺德,治疗领域主要为内分泌与代谢、神经、泌尿、心血管系统。6款药物均靶向肝脏,并进行了化学修饰。除了Onpattro递送方式为LNP(脂质纳米粒),并且给药方式为静脉输注外,其它药物的递送系统均为GalNAc,给药方式均为皮下注射,详见表1。
表1六款siRNA上市药物(截止2024.1)
由于siRNA自身的物理化学特性,未经修饰的siRNA给药后不仅会很快被机体清除,同时还有脱靶毒性的风险。因此,需要同时借助化学修饰和合适的递送系统来达到治疗目的。已上市的6款siRNA药物均使用了化学修饰和递送系统。第一代化学修饰是对磷酸骨架的修饰,如硫代磷酸酯(PS)。第二代化学修饰是核糖修饰,其中2-O-甲基(2-OMe)、2-O-甲氧基-乙基(2-MOE)和2-氟(2-F)修饰是最常用的类型。第三代化学修饰为核糖五元环改造。经过化学修饰后,共同的特点是增加了siRNA的稳定性,但同时也存在一些缺点,如经PS修饰后降低与靶基因结合的亲和力,与某些蛋白的非特异性相互作用可能引起细胞毒性。
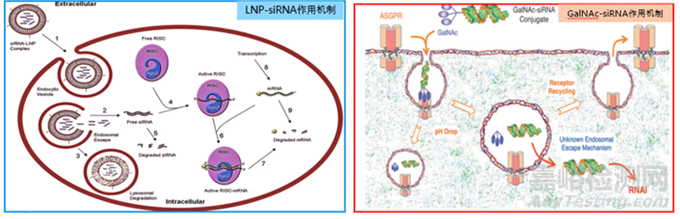
单纯通过化学修饰还是很难使siRNA到达靶部位。因此,递送系统是寡核苷酸药物研发中成功的关键因素,也是药物研发公司的技术护城河。目前已上市的6款siRNA药物,只有Patisiran递送系统是LNP(图1左),其余的递送系统均为GalNAc(图1右)。LNP主要由4部分组成:可电离脂质、胆固醇、辅助磷脂、PEG-脂质。其中,可电离脂质是最关键的辅料,它在不同的pH条件下表现出不同的带电特征,酸性pH值条件下带正电荷,而在生理pH条件下基本呈中性,可离子化特性为siRNA跨越生物屏障提供了智能化的保护。5款GalNAc-siRNA偶联物均使用L96作为载体,该结构由三部分组成,包括三触GalNAc靶头、连接臂以及siRNA分子。GalNAc以三价态的方式共价偶联至核酸3’末端,形成GalNAc-siRNA偶联物。GalNAc是去唾液酸糖蛋白受体(ASGPR)的配体,ASGPR在肝细胞的膜表面高度特异性表达。GalNAc-siRNA偶联物通过特异性结合ASGPR,在内吞作用下将siRNA从细胞表面转运至细胞中。随后GalNAc-siRNA偶联物与ASPGR分离,ASPRGR回到细胞表面,而GalNAc-siRNA偶联物进一步解离,释放出的游离siRNA在细胞质中沉默基因以发挥药效。
图1 递送系统LNP, GalNAc及作用机制
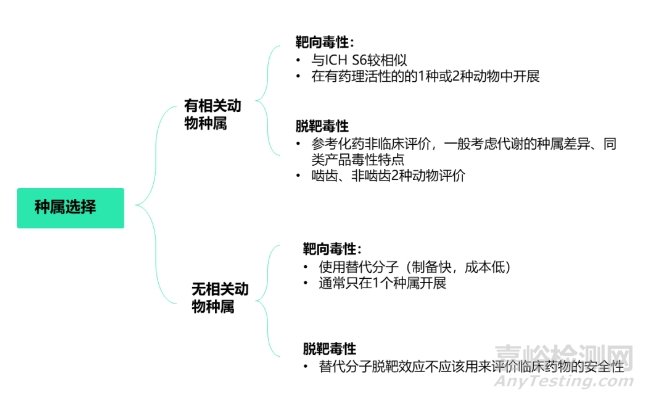
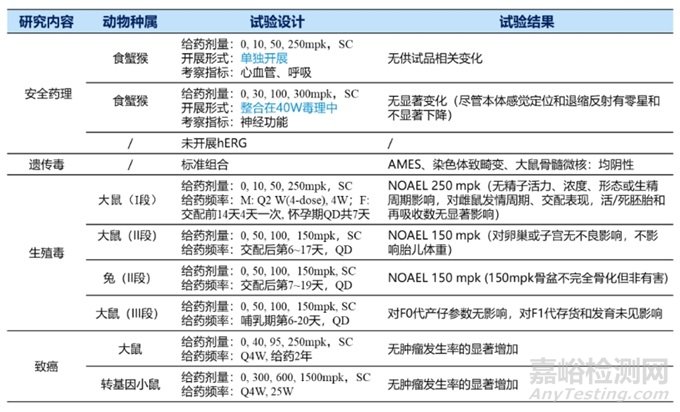
siRNA药物化学合成药物,虽具备生物制品属性,但按新化学实体监管。所以不仅在科学认知上存在一些缺口,也存在一些监管指南空缺。目前中国尚无相关指导原则。除了ICH系列指导原则外(ICH S6/M3/S7/S2/S8),日本PMDA 2020年发布了《寡核苷酸治疗产品非临床安全性评价指导原则》可供参考;FDA也发布了用于工业界的相关指导原则,如《寡核苷酸治疗药物开发的临床药理学考虑》等。根据日本PMDA发布的《寡核苷酸治疗产品非临床安全性评价》指导原则,siRNA药物需要2个种属进行临床前安全性评价,啮齿类为大鼠或者小鼠,非啮齿类一般为食蟹猴。目前已上市的6款siRNA,非啮齿类均是食蟹猴,啮齿类分别为大鼠(5款)、小鼠(Nedosiran)。非啮齿动物选择猴的主要原因如下:猴和人的目标mRNA具有高度或完全同源性,而且猴种属可以更好地评估siRNA放大的药理学效应,比如Inclisiran核苷酸序列与人和猴PCSK9中的类似序列完全同源,且用猴开展药效。siRNA药物临床前评价动物种属选择见图2。
图2 siRNA药物临床前评价动物种属选择
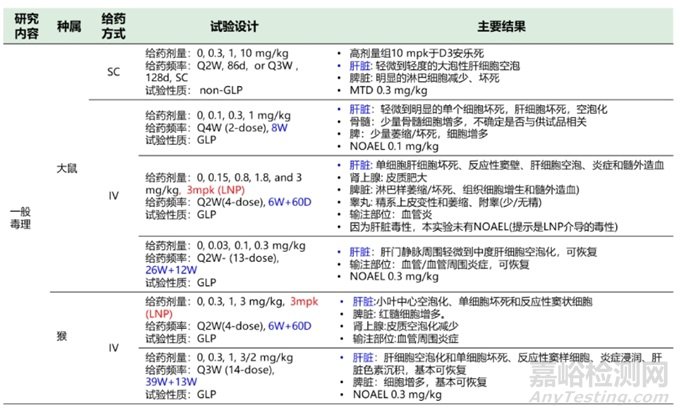
根据已上市的6款siRNA药物申报资料,临床前开展的安全性评价试验总结如表2。
表2六款siRNA药物临床前安评试验总结
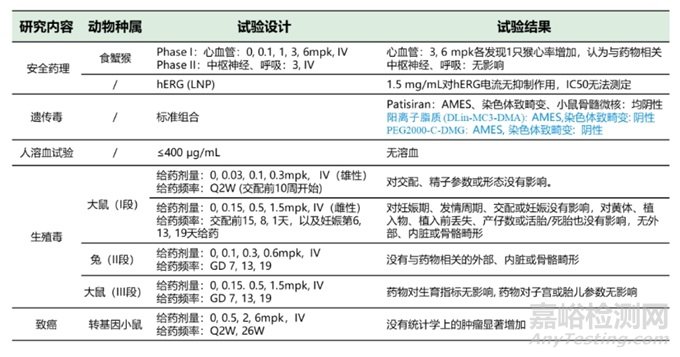
考虑到已上市的6款siRNA药物临床前毒理数据较多,仅以递送系统为LNP的药物Patisiran,递送系统为GalNAc的药物Inclisiran为代表,分析临床前安评结果。Patisiran是Alnylam研发,于2018.8 FDA批准上市的首款siRNA药物。适应症为遗传性转甲状腺素蛋白淀粉样变性(hATTR)引起的周围神经病变疾病,靶点TTR(转甲状腺素蛋白)基因。临床给药为静脉滴注,3周一次, 0.3 mg/kg。递送载体为LNP。作用机制是通过RNA干扰引起突变型和野生型TTR mRNA的降解,从而导致血清TTR蛋白和组织中TTR蛋白沉积的减少,达到治疗疾病的目的。Patisiran临床前安评结果如表3。
表3 Patisiran临床前安评总结(备注:如26W+12W表示26周给药,伴随12周恢复期)
表3 Patisiran临床前安评总结(续)
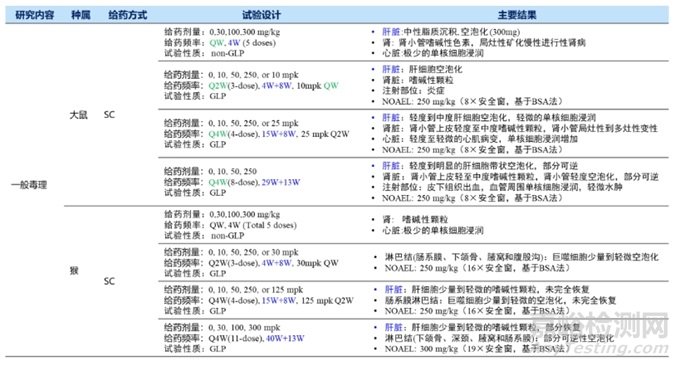
Inclisiran研发公司Novartis/Alnylam,于2020.12欧盟首次获批,全球首个siRNA降胆固醇疗法药物,适应症原发性高胆固醇血症或混合性血脂异常,靶点PCSK9。临床给药途径为皮下注射,第0、3个月各给药一次后,维持期6个月一次,一年只需2次。递送系统为GaINAc,作用机制是与编码PCSK9蛋白的mRNA结合,通过RNA的干扰作用来防止肝脏生成PCSK9蛋白,从而达到降低血脂的目的。Inclisiran临床前安评结果总结如表4。
表4 Inclisiran临床前安评总结
表4 Inclisiran临床前安评总结(续)
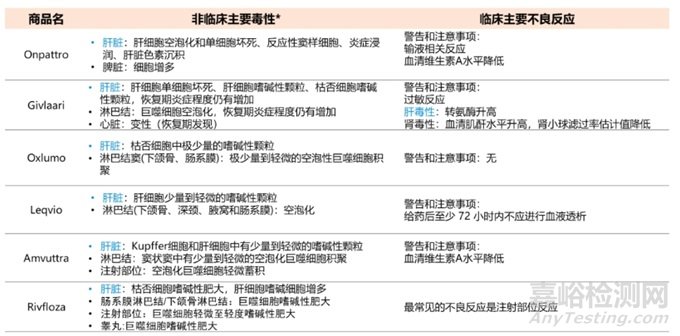
为了探究已上市的6款siRNA非临床毒性和临床主要不良反应是否有关联性,通过整理支持NDA的NHP长毒试验毒性结果,以及临床警告和注意事项,得出非临床和临床关联性较强的是肝脏毒性,需要关注血生化一些指标变化,如ALT, AST,可在临床阶段进行密切监控。具体情况见表5。
表5 六款siRNA药物非临床和临床毒性关联性分析(*:数据来源于支持NDA的NHP长毒试验)
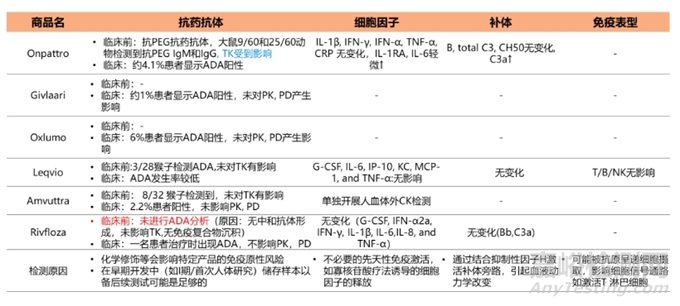
因为siRNA药物具备大分子药物属性,所以也需要进行免疫毒性考察。对已上市的6款siRNA药物免疫毒性进行总结,如抗药抗体(ADA)、细胞因子(CKs)、补体、免疫表型(IPT)。具体结果见表6。
表6六款上市siRNA药物免疫毒性总结(“-”表示未披露)
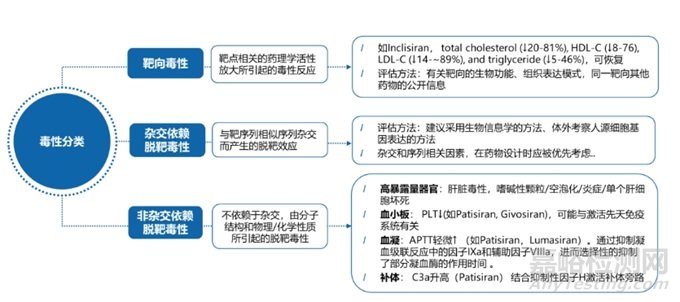
理论上讲,若siRNA和靶mRNA准确地结合在一起,则可成功的沉默靶基因。然而,许多siRNA都存在脱靶,并且大多数siRNA分子可能并不具有靶向性。siRNA的引入可导致脱靶效应,即抑制目标基因以外的其他基因,导致非靶基因表达突变的意外后果。根据日本药品医疗器械管理局(PMDA)发布的《寡核苷酸治疗产品非临床安全性评价指导原则》,寡核苷酸的毒性分析分为靶向毒性、脱靶毒性,详细见图3。
图3 siRNA药物毒性分类
肝脏是siRNA药物最关注的器官。关于肝脏毒性,通过总结已上市的6款siRNA药物,将从药物特点、药代、病理进行综合性分析。药物特点方面,6款siRNA药物靶点均与肝脏相关,在肝脏中合成或表达;均进行了化学修饰,如经硫代磷酸脂修饰后可能导致高蛋白结合的肝毒性;递送系统无论是LNP或GalNAc均有肝靶向性。目前LNP-siRNA药物Patisiran静脉给药后80%~90%的LNP会进入肝脏,最终被肝细胞吸收;GalNAc-siRNA经ASGPR介导发生细胞内吞而进入靶组织,ASGPR在肝细胞上大量表达。药代方面,组织分布结果均显示肝脏浓度最高,如Lumasiran猴10mg/kg时,AUC高达76700h*μg/mL,且消除半衰期长,高达409h。用于支持NDA的猴长毒试验中,组织病理均发现肝脏变化,从轻到重依次是:(1)肝细胞/枯否细胞嗜碱性颗粒:可能为供试品或其代谢产物,也可能是细胞摄取寡核苷酸的适应性改变;(2)肝细胞空泡化:可能为继发于胞浆内嗜碱性颗粒蓄积的改变;(3)炎症浸润:高浓度药物分布时,会激活促炎性通路(4)肝细胞单细胞坏死:高剂量下嗜碱性颗粒大量蓄积时可观察到病理损伤。此外,大部分药物还伴随血生化临检指标变化,如ALT, AST升高。组织的病变是否为有害变化,要根据组织本身的损伤,相关指标的变化,可恢复情况,以及其它的siRNA药物是否有类似改变等综合判断。
除了肝毒性,序列相关引起的脱靶毒性还有免疫刺激反应,其毒性在很大程度上取决于核苷酸序列设计和化学修饰。siRNA药物通过激活先天免疫系统,导致注射部位反应(如Patisiran,Inclisiran)、血小板(PLT)减少(如Patisiran, Givosiran)。免疫刺激评估,除了体内试验相关指标的检测,也可在体外用分离的外周血单核细胞或全血测定预测细胞因子释放来评估。
非杂交相关的脱靶毒性,如凝血功能改变,siRNA药物通过抑制凝血级联反应中的因子IXa和辅助因子VIIIa,进而选择性的抑制了部分凝血酶的作用时间,从而以一种与杂交和序列无关的方式抑制凝血(如Patisiran,Lumasiran APTT轻微升高)。此外,siRNA药物还可以结合抑制性因子H激活补体旁路,补体过度激活可使补体功能改变,引发继发性炎症和血管炎等(如Patisiran C3a升高,发现肝脏炎症浸润、输注部位血管周围炎症)。
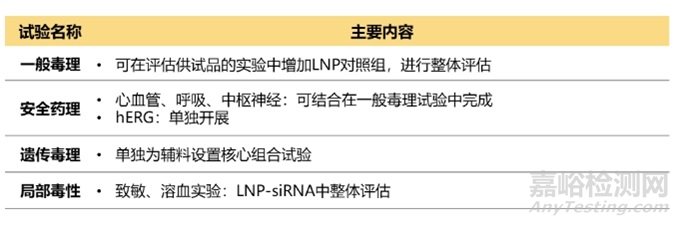
除了关注siRNA药物本身外,还要关注递送系统毒性,如LNP。LNP含有新的辅料,或成分比例发生了改变,需要对LNP进行考察。虽然没有专门针对LNP的指导原则,但可以参考相关指导原则,如《新药用辅料非临床安全性评价指导原则》、《纳米药物非临床安全性研究技术指导原则》、《Guidance for Industry : Liposome Drug Products Chemistry, Manufacturing, and Controls; Human Pharmacokinetics and Bioavailability; and Labeling Documentation》等。此外,还可以参考Patisiran关于LNP的考察。若LNP是按非单独辅料注册标准申报,且考虑到siRNA需要两种动物种属,可按与非临床安全性评价中相同的动物种属伴随或单独考察LNP。IND申报LNP需要开展的毒理试验推荐见表7。
表7 LNP毒理IND评价(非单独辅料注册标准)
据报道,siRNA药物发展方向依然主要集中在递送系统、修饰、脱靶效应、适应症拓展方向,且相辅相成。比如在递送系统方面,Alnylam开发了一种连接到siRNA的短脂质链——C16偶联物,使其能被多种类型细胞吸收(CNS,肺,眼等),从而拓展了siRNA类药物适应症;新的递送平台GalAhead™和PDoV-GalNAc提供了更高的递送效率。在修饰方面,例如增强稳定性化学(ESC),很多在研项目已经开始使用了。关于脱靶应在药物设计时就考虑,而不是通过毒理评估。如Alnylam公司对siRNA脱靶预测,淘汰不符合要求的siRNA序列。
2018年,第一款siRNA药物Onpattro的批准上市对siRNA药物领域产生了重大影响,标志着诺贝尔奖成果已从理论概念转变为实际治疗用途,这是一个引人注目的里程碑。截至2024年1月,已有6款siRNA药物上市。通过对这6款已上市siRNA药物临床前安评总结分析,可以为此类药物临床前毒理设计提供参考。相信将来会有更多的siRNA药物上市,而相关的临床前指导原则也将随之逐渐完善和发布。
【参考文献】
寡核苷酸治疗产品非临床安全性评价指导原则,日本药品和医疗器械管理局(PMDA),2020.03
王恒,李华,汪溪洁,等. 小核酸药物非临床特点和药理毒理评价策略[J]. 中国新药杂志,2022,31(12):1137-1145. DOI:10.3969/j.issn.1003-3734.2022.12.001
余珊珊、胡晓敏、王海学等. 治疗用单链寡核苷酸药物的非临床研究评价概述. 中国新药杂志, 2018, 27(10): 1122-1129.
周宇,王士奇,孙涛, 等. 日本药品和医疗器械管理局《寡核苷酸治疗产品非临床安全性评价指导原则》指南介绍. 中国临床药理学杂志, 2022, 38(22): 2788-2792
Clinical Pharmacology Considerations for the Development of Oligonucleotide Therapeutics Guidance for Industry, FDA, 2022.06
Agrawal, S. & Kandimalla, E. Role of Toll-like receptors in antisense and siRNA [corrected]. Nature biotechnology 22, 1533-1537, (2004).
Henry, S. et al. Activation of the alternative pathway of complement by a phosphorothioate oligonucleotide: potential mechanism of action. The Journal of pharmacology and experimental therapeutics 281, 810-816 (1997).
Sheehan, J. & Phan, T. Phosphorothioate oligonucleotides inhibit the intrinsic tenase complex by an allosteric mechanism. Biochemistry, 40(16), (2001).
Andersson, P. Preclinical Safety Assessment of Therapeutic Oligonucleotides. Methods in molecular biology (Clifton, N.J.) 2434, 355-370, (2022).